May 4, 2016

May 4, 2016
 For those of you unaware, the FDA submitted a draft guidance on data integrity for comment in April. This is significant in that the last formal written guidance provided from FDA on this subject matter was the Part 11 Scope and Application Guide that was published in August 2003.
For those of you unaware, the FDA submitted a draft guidance on data integrity for comment in April. This is significant in that the last formal written guidance provided from FDA on this subject matter was the Part 11 Scope and Application Guide that was published in August 2003.
Relative to this new guide on data integrity, I have summarized in this article what I consider are the top takeaways that one should keep in mind as you look to evaluate your own internal processes around data integrity.
Takeaway #1: Data Integrity Definition
The term Data Integrity is actually defined: The simple definition is reliable and accurate. The detailed definition is having the ALCOA attributes. The FDA also lays the context around this thinking by identifying existing regulations/requirements that speak to the ALCOA attributes including the following parts: 211.68, 211.100(b), 211.160, 211.180, 211.188, 211.194, and 212.60(g).
Takeaway #2: Audit Trail for a Record
The definition of the term Audit Trail has been further clarified to specifically apply to a record, not just a system. This reinforces the concept of traceability of “who, what, when, and why” actions to a given record, not just a time-based log of user actions within the system.
Takeaway #3: Audit Trail Review Timing
Also, related to audit trails, expectations were further clarified that the audit trail for a given critical data record should be reviewed before final approval of the record. Periodic or routine scheduled audit trail review should be based on the complexity of the system and intended use. In addition, specific audit trail meta data categories that should be included in review of critical data are the following:
a. The change history of finished product test results
b. Changes to sample run sequences
c. Changes to sample identification
d. Changes to critical process parameters
Takeaway #4: Who Should Review Audit Trails
Since audit trails are considered part of the associated records, personnel responsible for record review under cGMP should review the audit trails that capture changes to critical data associated with the record as they review the rest of the record. For example, all production and control records, which includes audit trails, must be reviewed and approved by the quality unit (§ 211.192).
Takeaway #5: Access Restriction / Separation of Roles
FDA suggests that the system administrator role, including any rights to alter files and settings, be assigned to personnel independent from those responsible for the record content. If these independent security role assignments are not practical, FDA recommends documented alternate control strategies such as second person review of settings and content.
Takeaway #6: Training
Finally, FDA reiterates the importance of training personnel to understand and detect data integrity possibilities and issues. Human error tends to be the biggest contributing factor relative to data integrity issues. Never assume it won’t happen to your organization or that one-time training is enough. Supporting programs need to be in place, maintained, and continuously improved.
For those of you interested in giving FDA your feedback, comments on the new FDA guide can be submitted here.
Learn more about ProPharmaCSV services.
Contact us to get in touch with our subject matter experts for a customized Computer Systems Validation presentation.
February 24, 2016
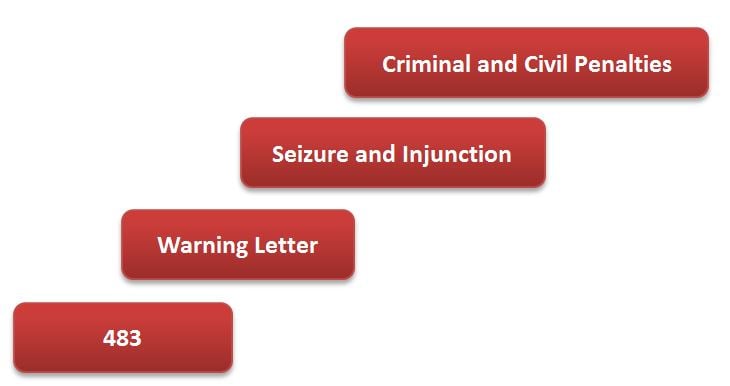
Starting with the issuance of a 483, the stepwise FDA enforcement process can be illustrated as follows: Given the seriousness of a Seizure and Injunction scenario, not to mention potential jail time...
February 25, 2016
On Thursday, February 25th, FDA announced plans to hold a public workshop to discuss the evidentiary standards necessary to support biomarker qualification. The workshop, entitled “Developing an...
March 24, 2021
A combination product is composed of any combination of a drug and a device; a biological product and a device, a drug and a biological product, or a drug, device, and a biological product. Consider...