July 12, 2021
July 12, 2021
Before using a significant risk medical device in a clinical study, Sponsors must first obtain FDA approval. Frequently, the request for an investigational device exemption (IDE) is disapproved, and the impact of this disapproval can be significant, as Sponsors have invested a great deal of time and resources into development of the device before they get to this stage. However, disapproval is not the end of the process. Sponsors can request a Submission Issue Meeting in which they can address the FDA’s concerns and elicit input from the Agency which will help ensure that a resubmitted IDE application is approved.

All clinical investigations conducted in the US involving a significant risk medical device must have an FDA-approved IDE. An IDE allows Sponsors to use an investigational device for the express purpose of collecting clinical data. Once an IDE approval is granted by the FDA, a clinical study can commence and data can be collected to support approval or clearance of a device marketing application (e.g., premarket approval (PMA), 510(k)).
The Sponsor of the clinical trial is responsible for submitting the IDE application to the FDA and obtaining IDE approval. This application is a data package that is analogous to an investigational new drug application (IND) for drugs. The intent of the application is to demonstrate to the FDA that the study design is sound, the subjects receive adequate protection, and that the device is safe enough to conduct a clinical study under the IDE.
There are no formal forms for an IDE application, which makes the process particularly challenging. However, there is certain information that an IDE application must include. In the application, the Sponsor must demonstrate that there is no reason to believe that the risks to human subjects from the proposed investigation are outweighed by the anticipated benefits to subjects. The Sponsor must also demonstrate the importance of the knowledge to be gained, that the investigation is scientifically sound, and that there is reason to believe that the device, as proposed for use, will be effective.
After an IDE application is submitted, FDA has 30 days to review it, after which the Agency can respond in one of three ways:
If a conditional approval or outright disapproval is received and the Sponsor has significant questions or wants to discuss the content of the deficiencies outlined in the letter, the Sponsor can request a Submission Issue Meeting with the FDA. This request must be submitted within 30 days, and the Agency aims to put the meeting on their calendar within 21 days of receiving the request to discuss the matter.
A Sponsor can consider the Submission Issue Meeting a success if they leave the meeting with the feedback they need to re-submit a successful application. This meeting provides an excellent opportunity to gain clarity from the Agency regarding its concerns. In addition, it’s also an opportunity for the Sponsor to present their thinking to FDA in support of to the application, address FDA’s concerns, and get an initial take on the probability of FDA approving a resubmitted application.
The success of the meeting is based on how well a Sponsor prepares in advance. Sponsors typically only get one hour with the FDA during a Submission Issue Meeting. As such, the first thing you should do is prioritize what issues you want to discuss. The issues that the Agency has suggested could put a significant hold on the IDE should take precedence. It is also important that you plan for and prioritize how you plan to respond to these issues. Since the goal is to get advice from the Agency, your presentation should be designed to elicit helpful feedback. The overall objective of the meeting is to gain as much clarity as possible about whether a resubmitted application will be approved or disapproved. Leaving the room with any ambiguity is the hallmark of an unsuccessful meeting.
One major benefit to having ProPharma’s team of experts at your side prior to and during the Submission Issue Meeting is the know-how and experience we bring in successfully leading IDE applications to approval using this mechanism. Our staff are highly experienced and have worked extensively with FDA to achieve positive outcomes through the IDE application process. Knowing how FDA thinks and being able to get to the heart of their concerns translates to devising and presenting plans in the meeting that the Agency can agree to. We excel in this area and can provide this unique insight to make sure that your Submission Issue Meeting after initial IDE disapproval is a success.
Do you want ProPharma’s assistance with your IDE submission? Interested in having ProPharma help you prepare for and attend your Submission Issue Meeting with the FDA? Contact us today to learn how we can help with all aspects of medical device regulation.
TAGS: Medical Devices fda submission FDA Meetings Regulatory Sciences
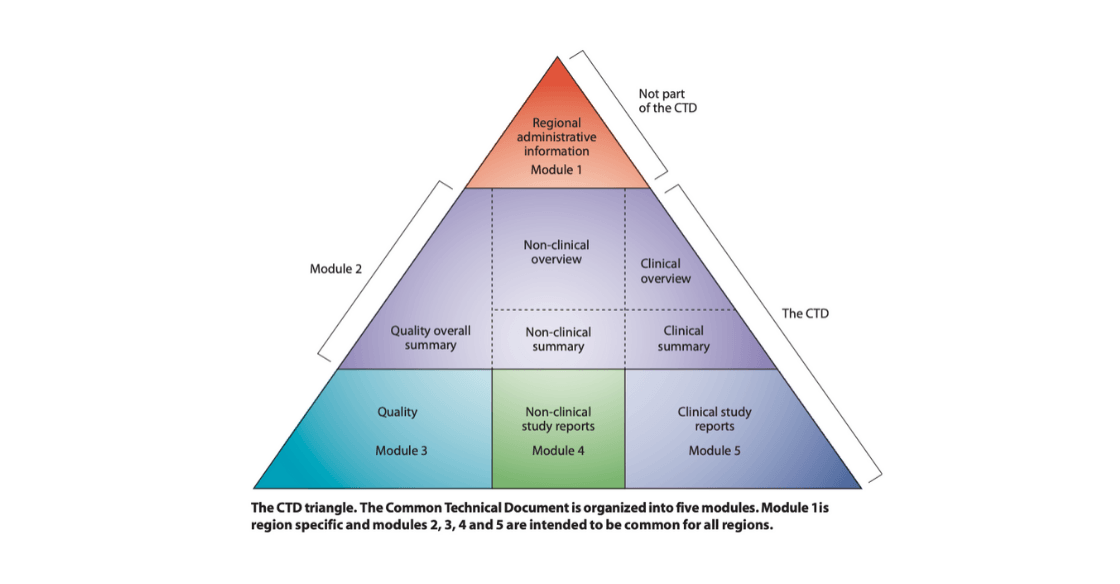
February 5, 2020
The Importance of the eCTD To harmonize the process of regulatory reviews for global drug development, a unified structure was developed and implemented for electronic Common Technical Document...
May 14, 2015
In a final guidance published in the Federal Register on May 5, FDA laid out the rules and specifications for providing regulatory submissions in electronic format. Perhaps more important than the...

August 2, 2022
Guidance Document August 2022 This guidance provides instructions for the electronic submission of expedited individual case safety reports (ICSRs) from investigational new drug (IND)-exempt...