The Importance of the eCTD
To harmonize the process of regulatory reviews for global drug development, a unified structure was developed and implemented for electronic Common Technical Document (eCTD) submissions. The structure is based on the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines.
The Food and Drug Administration (FDA) encourages the use of ICH guidelines as the backbone and preferred format for submissions in the United States.
In May 2017, the agency announced the eCTD structure would be the only accepted form of submissions for NDAs, ANDAs, and BLAs. As of May 2018, INDs are also required to follow the eCTD submission structure.
Advantages of the eCTD Structure
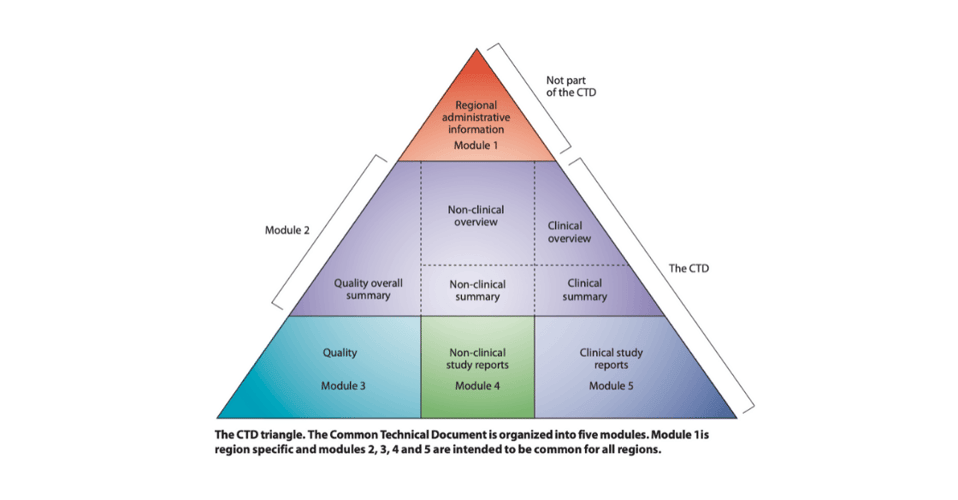
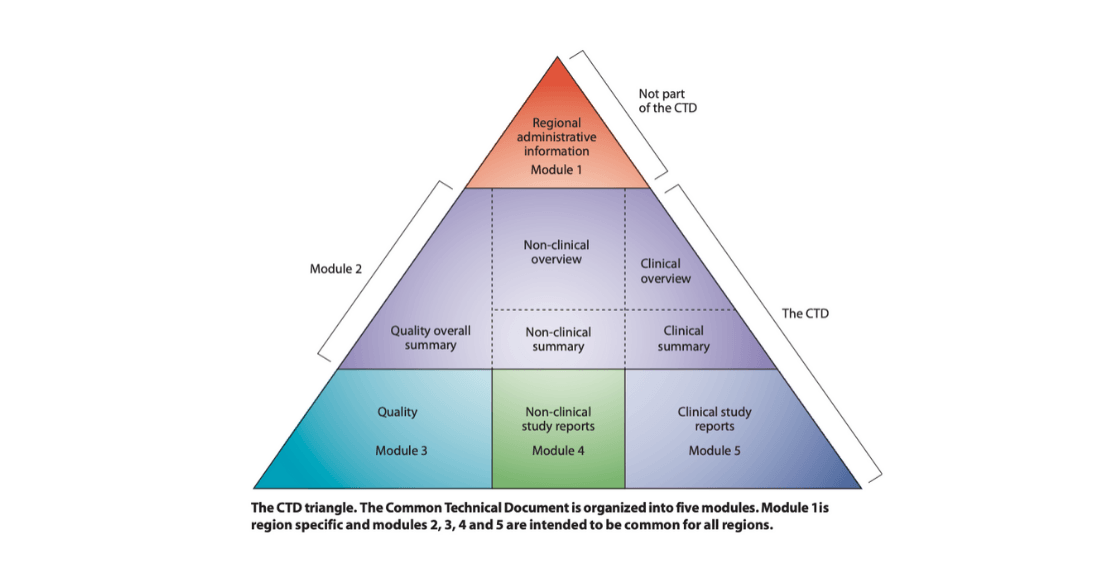
The CTD comprises 5 modules, each with its own set of requirements in terms of content and documents.
Advantages of the eCTD structure:
- Reviewers are already familiar with the content and document standards
- Local affiliates can review updates in real-time
- FDA reviewers can review faster and more efficiently, shortening time to approval
- Handling, managing, and archiving trial and document-essential information is less time-consuming
- Documents are easily accessible via search and tracking
- The risk of missing or duplicated documents is minimized
Challenges of the eCTD Structure
While most larger organizations have taken proactive measures to fully implement eCTD submissions, smaller companies may not have fully developed measures to ensure coordination of documents across the eCTD. As a result, challenges can arise.
Below are 3 of the obstacles that companies may face in preparing for an electronic submission:
Transitioning from paper to eCTD
All organizations are now required to provide an electronic format for submissions; for smaller companies in various stages of submission planning, paper documents may still need to be converted into a digital format.
Transitioning to electronic filing can represent a major undertaking for companies that have not been actively preparing for the last few years. ProPharma can help streamline the conversion process to ensure full eCTD-compliance.
Ensuring submission-readiness
To successfully migrate to an electronic submission platform, companies must be equipped with cutting-edge tools such as:
- eCTD publishing software
- FDA fillable forms
- Electronic Submission Getaway (ESG)
- Viewing software
Each tool requires some form of selection, setup, validation, compatibility checks, training, maintenance, and software upgrades.
Following guidance and directives for a successful submission
Despite ICH guidance and FDA directives, errors and mishandling can occur, leading to rejection of a submission.
The agency lists top 3 reasons for rejection:
- Use of duplicate sequence number
- Lack of consistency between eCTD backbone files and regulatory forms
- Lack of conformity with the published FDA Data Standards catalog
If you lack in-house technical expertise, now is a good time to bring in outside support. The move to a fully digital submission process requires effective coordination and communication of documents across many functional areas. ProPharma has a proven track record of delivering project management solutions for eCTD submissions, as well as in many other areas, including Regulatory, Quality, and IT.
Navigating Regulatory Requirements for Successful Submissions
Organizations that partner with experts on local and global regulations and requirements will gain a significant competitive edge on the way to a successful submission.
Contact ProPharma for a no-cost Regulatory Strategy Consultation today. Let us guide you through the eCTD process and partner with you to manage the multitude of requirements associated with success.
TAGS: Abbreviated New Drug Application (ANDA) New Drug Application International Council for Harmonisation (ICH) Biologics License Application (BLA) electronic Common Technical Document (eCTD) Clinical Research Solutions

