
Laying the Groundwork for ANDA Success: The Role of the 505(j) Pathway and Reference Products
Identifying issues early in the ANDA development process is key to ensuring a timely review and overall successful ANDA application. The 505(j) pathway is what allows for a generic drug to be reviewed and ultimately make it to the US marketplace. However, using the "same as" principle will only get an applicant so far as the complexity of new sponsor NDA products becomes increasingly complex.
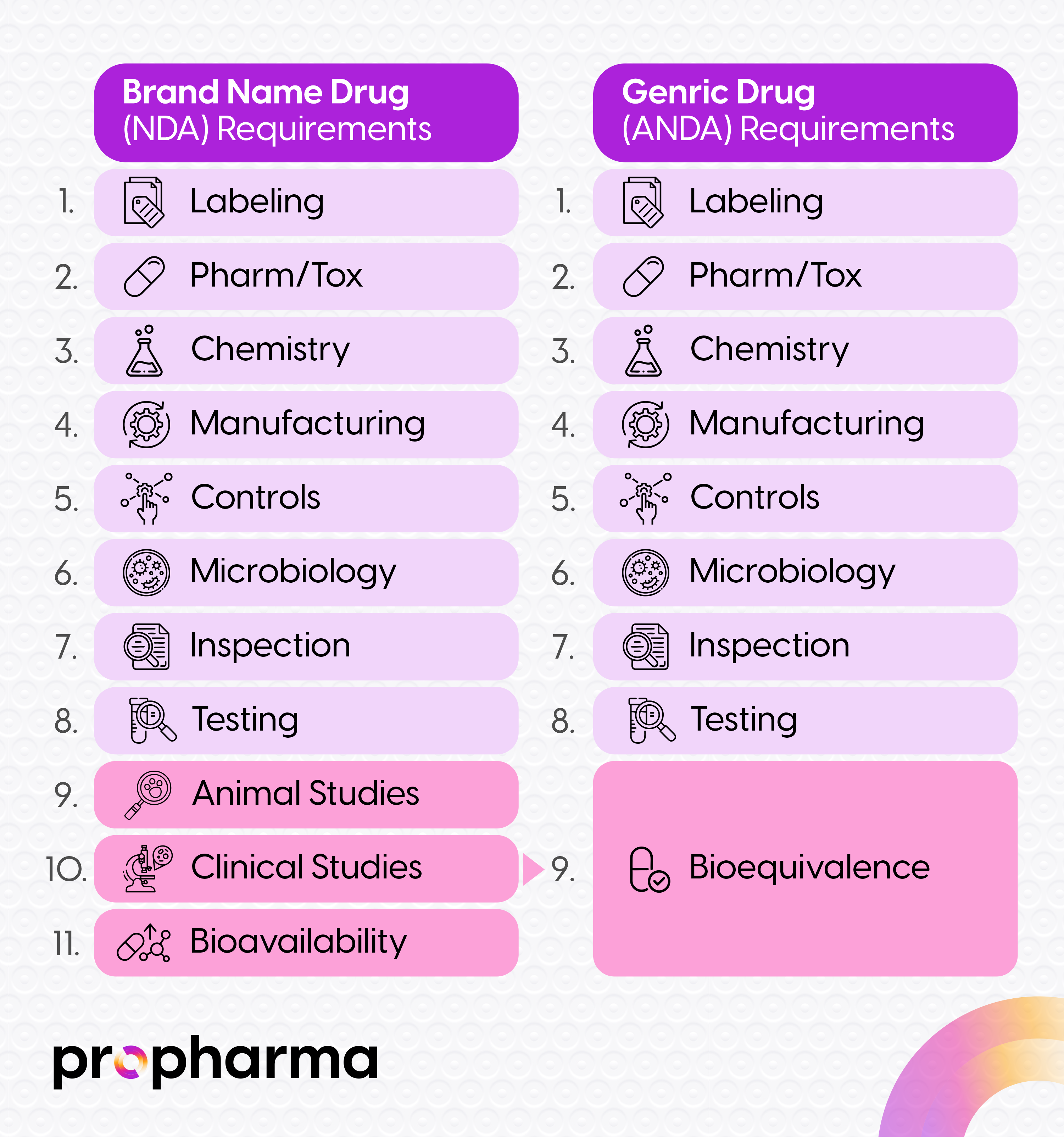
An ANDA application relies on the initial safety and efficacy findings of the NDA product which is considered the basis of submission (from here on the reference product). The ANDA establishes bioequivalence with respect to the reference product and is then allowed to utilize all the clinical data for safety and efficacy thus not having to complete their own clinical studies. The ANDA applicant then must adhere to regulations and FDA guidance documents to ensure they are adequate with their quality and administrative information that includes labeling.
To provide deeper insight into the complexities of ANDA development, former FDA reviewer, Marshall Florence, PharmD, Vice President of Labeling and Strategy at ProPharma, shares practical guidance on how reference product selection shapes the foundation of a successful submission. In this Q&A, he explores why identifying the appropriate basis of submission is critical to ensuring regulatory alignment, minimizing review cycles, and achieving timely approval.
What is the importance of selecting the basis of submission for an ANDA?
Dr. Marshall Florence: Understanding the requirements and allowable differences is a key component to ANDA development, market strategy and overall business operations. For example, if a company wants to develop a generic version of vancomycin hydrochloride there are a few things to understand when creating the ANDA package. Initial questions in development and planning would include:
What is the NDA reference product that will be relied on for the creation of quality information and labeling?
Dr. Marshall Florence: In this example, there are numerous products listed and some have different requirements than others. NDA 208910 for Firvanq is a newer formulation of vancomycin hydrochloride and has vastly different requirements than NDA 213895 for vancomycin hydrocholoride. Although the active pharmaceutical ingredient (API) is the same including the salt formulation, the products are extremely different.
Firvanq, is a newer product that comes as a kit and has specific instructions with the included diluents and use of the product compared to the older NDA oral suspension formulation. Although they are both used for the same indications, they have different places in the commercial marketplace.
ANDAs that choose Firvanq as their basis of submission must ensure their quality data matches that of that specific NDA along with their labeling. The product would need to include the diluents, proper packaging of the kit with respect to the reference product and ensure their labeling is the "same as" Firvanq while also navigating any patents and exclusivities listed in the OrangeBook for that NDA. This is a much more complex process compared to ANDA products that choose the NDA for "vancomycin hydrochloride for oral suspension" which is a simpler package and label and older product with no patents or exclusivities.
Additionally, ANDA applicants can leverage the OrangeBook to see market competition for approved products and determine their barriers to entry and commercial marketing plan for their product.
This is only one example of a potential issue for product selection and product specific issues and this type of issue can be found across numerous NDA products and can encompass other regulatory aspects such as patents and exclusivities, USP requirements, and other labeling issues.
What happens if I have a question during development prior to submitting an ANDA?
Dr. Marshall Florence: Let's say I have questions in developing the regulatory plan for an ANDA, how would I get answers or insight into my product?
First, you can leverage the experience and professional insight of ProPharma which includes numerous FDA veterans, (like myself), that know the innerworkings of the Agency to help you build a successful game plan.
There are other ways that you can look to figure things out based on other ANDA approvals with respect to labeling issues, product issues like pre-filled syringes, applicators, or other drug-device issues. Another way that you could attempt to get information or official Agency guidance would be through a controlled correspondence (CC). An applicant can submit a CC ahead of time to gain information from FDA if it is not a review specific issue. Often times, the phrasing of the question and supporting information will trigger the CC to be answered with a response citing that the question is deemed as a review issue and would be reviewed and commented on during a full review. It is important to realize how issues should be asked and what information is important for the Agency to comment on that will impact your overall submission.
My basis of submission has several patents and use codes listed in the OrangeBook, how do I craft my labeling effectively?
Dr. Marshall Florence: Looking at the OrangeBook for patent and exclusivity information can sometimes be an easy to-do item while other times it proves to be a lengthy chore that can confuse even the most seasoned reviewer or regulatory affairs expert. In the earlier scenario, older drugs often have nothing listed and can be very easy to navigate while newer therapies have a multitude of protections that affect different sections and even sentences within the labeling. Understanding how patents and their associated protections, along with exclusivities, is a key aspect to submitting a quality ANDA labeling submission in addition to the required patent certifications and exclusivity statements.
First and foremost, understanding and creating the patent certification and exclusivity statement documents as required is step one. If not done correctly, this can cause costly delays to the overall application timelines. Ensuring that paragraph IV certifications are documented and patent owners are notified with delivery receipts are a requirement to ensure delays are not incurred during the review process. If a certification is switched, understanding that re-notification is required is also key, as this can also impact review timelines.
Step two is ensuring that all labeling is adequately referenced to the appropriate coverage and correctly removed or retained based on the alignment and overall strategy for the patent certifications. If there is even one small discrepancy between the patent and exclusivity alignment or a discrepancy with the labeling, a deficiency will be issued. The issue may or may not be specifically addressed or a general deficiency will be issued, and it will be up to the applicant to identify and correct the disparity. This can lead to several cycles of resubmissions and reviews until the issue is fully rectified.
Another aspect that can cause confusion and add to review time involves pediatric information and exclusivity coverages with NPP and M codes and the removal of the information. FDA will issue a template for applicants to use for the appropriate carve out and disclaimer placement, but it can take months to receive from the Agency and add unnecessary review time while the applicants wait. An action will need to be taken on a 3-month clock upon submission of an applicant submitted carve-out labeling submission as opposed to a 3-month clock after receiving the FDA-created template and subsequent applicant labeling submission.
Understanding the process and being able to submit labeling that will mirror FDA's label template will effectively shorten ANDA review times and put you ahead of other generic competition.
Understanding how all of this works with respect to labeling is something the team of experienced regulatory consultants at ProPharma can do with ease to ensure that your submission is of the highest quality and overall reduces your deficiency comments and review timelines.
TAGS: Abbreviated New Drug Application (ANDA) Food & Drug Administration (FDA) FDA Submission Regulatory Sciences Labeling
