All Marketing Authorization Holders (MAH) of medicines for human use are facing what might feel for many, as a new requirement: review their drug products on the possible presence of nitrosamines.
The European Medicines Agency (EMA) has sent a notice to MAHs and adopted a scientific opinion concluding the review on nitrosamine impurities in human medicinal products, which were complemented with Q&A document. The Food and Drug Administration (FDA) issued a guideline for industry on Control of Nitrosamine Impurities in Human Drugs. Based on these guidelines EU marketing authorization holders are required to review their human medicinal drug products containing chemically synthesized and biological APIs on the potential risk of containing nitrosamines impurities. US MAHs must review medicines containing chemically synthesized APIs or APIs at risk due to other factors described in respective guidance.
MAHs are now facing a clear requirement with strict deadlines; fulfilling the three-step approach as outlined below:
Step 1: Risk Evaluation
MAHs should perform risk evaluation of their medicinal products. MAHs should prioritize products in order to establish the sequence in which their products are to be evaluated, based on the potential exposure of patients. MAHs should inform the concerned Competent Authorities when the risk evaluation is concluded. If a risk of the presence of nitrosamines is identified as a result of the evaluation, the MAH should proceed to Step 2.
Step 2: Confirmatory Testing
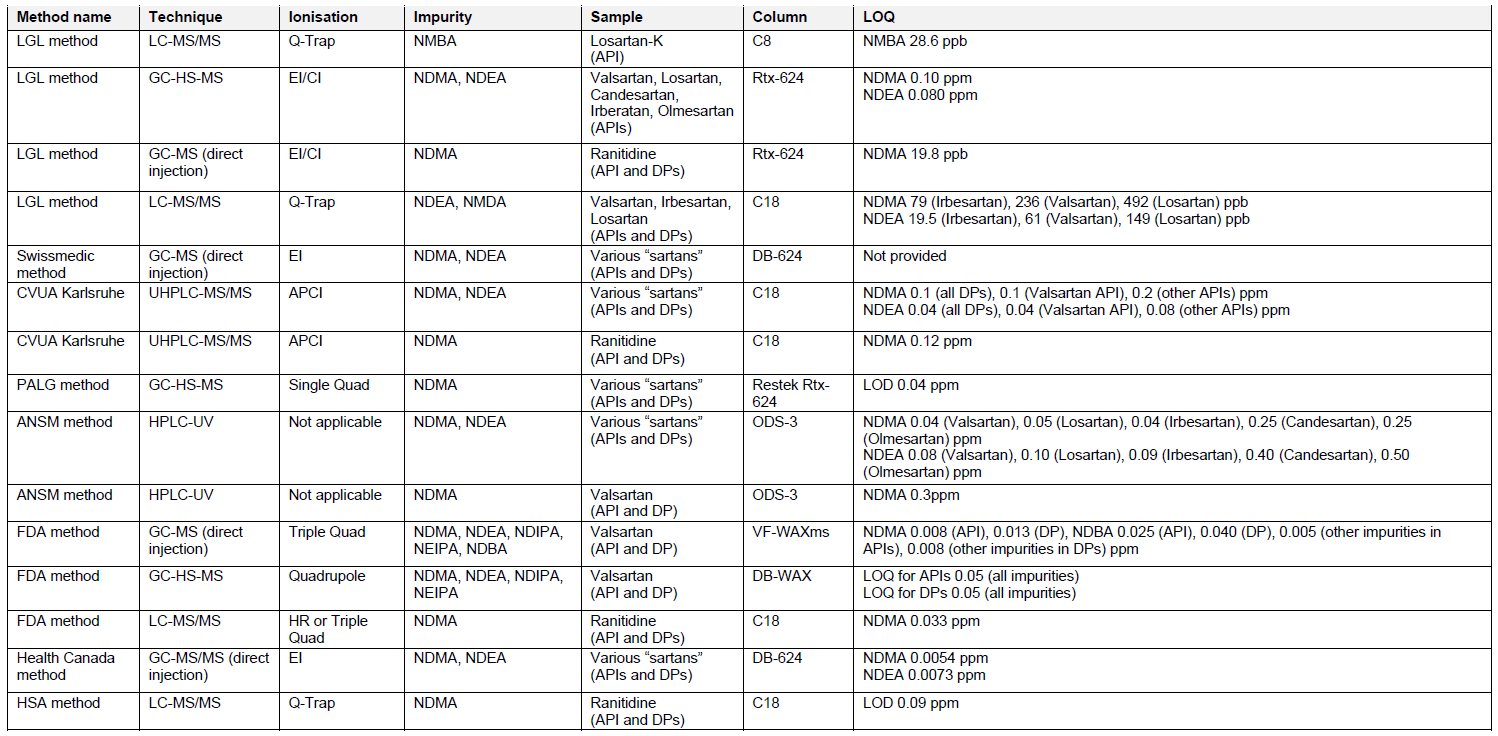
In the event that a risk of the presence of nitrosamines is identified as a result of the risk evaluation, confirmatory testing should be carried out using appropriately validated and sensitive methods in accordance with the prioritization based on the safety level of potentially affected patients. Products identified as high priority should be tested as soon as possible. Confirmatory testing of all medicinal products identified to be at risk of the presence of nitrosamines and submission of required changes in the manufacturing authorizations should be concluded as per applicable regional guidelines.
MAHs should inform the Competent Authorities immediately if tests confirm the presence of a nitrosamine impurity irrespective of the amount detected.
Step 3: Changes to the Marketing Authorization
MAHs should apply for a variation in a timely manner to introduce any required changes, such as an amendment of the manufacturing process or changes to product specifications.
At all steps, timelines should be shortened, and authorities immediately informed if findings indicate an immediate risk to the public health.
If for many pharmaceutical companies the three-step requirement does not present particular technical challenges, for all those situations where the portfolio of human medicinal products is large, the exercise can feel like an insurmountable mountain to climb.
FDA and EMA appeal to the MAH responsibilities as rationale for the strict implementation deadlines, explicitly reminding the MAH's that according to the existing legislation they are responsible for understanding of their own processes, which includes limiting the presence of unacceptable impurities. Furthermore, MAHs are obliged to develop and use suitable control methods to detect and quantify the impurities.
Other major regulatory bodies (e.g. Swissmedic, Health Canada) are also taking a position on nitrosamine impurities in medical products; positions that should be taken into account whilst making decisions for all the human medicinal products in a company’s portfolio worldwide. To better understand this global issue strong collaboration has been established among major international regulators. Therefore, regardless of the impact for companies, the main regulatory agencies around the world have issued clear statements, with specific deadlines, given the adopted standpoint that control of nitrosamines (as well as all impurities in general) already falls under MAH responsibilities and is therefore not a new requirement.
As the nitrosamine control issue does not formally fall into the category of new requirements, it still can prove to become a significant effort for all those pharmaceutical companies with a large portfolio of products; needing therefore close monitoring as to prevent product shortages. A multidisciplinary approach involving Formulation experts, Quality Assurance, Quality Control and Regulatory Affairs, is essential to fulfilling the new requirements and ensure control of nitrosamines presence in medicinal products for human use.