Since September 26, 2019, all EU Marketing Authorization Holders (MAHs) of medicines for human use are facing what might be regarded as a new requirement: review their drug products on the possible presence of nitrosamines. The associated risk evaluation should be finalized by MAHs before March 31, 2021 for chemical medicines and July 1, 2021 for biological medicines. In the US market, a separate due date of March 1, 2021 applies. In case of the identification of the risk of nitrosamine formation, MAHs should test drug products and APIs using sensitive and validated analytical methods.
Testing of chemical medicines identified to be at risk should be concluded at the latest before September 26, 2022, in EU and September 1, 2023 in US, three years after the publication of the guidelines. Additionally, testing of biological products should be conducted before July 1, 2023 for the EU products. Products identified as high priority should be tested as soon as possible. The outcome of the analysis of multiple products from different MAHs will be evaluated by Competent Authorities and will be used for investigating the root cause of nitrosamine formation.
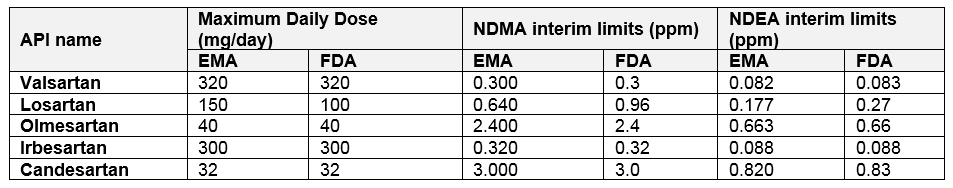
Based on the final legally binding decision of the European Commission, European Pharmacopea monographs of valsartan, candesartan, irbesartan, losartan, and olmesartan were revised setting limits for N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) given in Table 1. New monographs came into force on January 1, 2020. Similar interim specification limits were set by the Food and Drug Administration (FDA). Products containing NDMA or NDEA above these limits or containing both impurities at any level will have to be withdrawn from the market. After the transition period of 2 years, a strict limit of < 0.03 ppm for NDMA and NDEA will apply. Other nitrosamines which may be potentially present in “sartans” are e.g. N-nitrosoethylisopropylamine (EIPNA), N-nitrodiisopropylamine (DIPNA) and 4-[methyl(nitroso)aminobutanoic acid (NMBA). In April 2020 USP proposed the new general chapter <1469> on Nitrosamine Impurities providing the risk-based approach for the control of nitrosamines, based on the knowledge available until now.
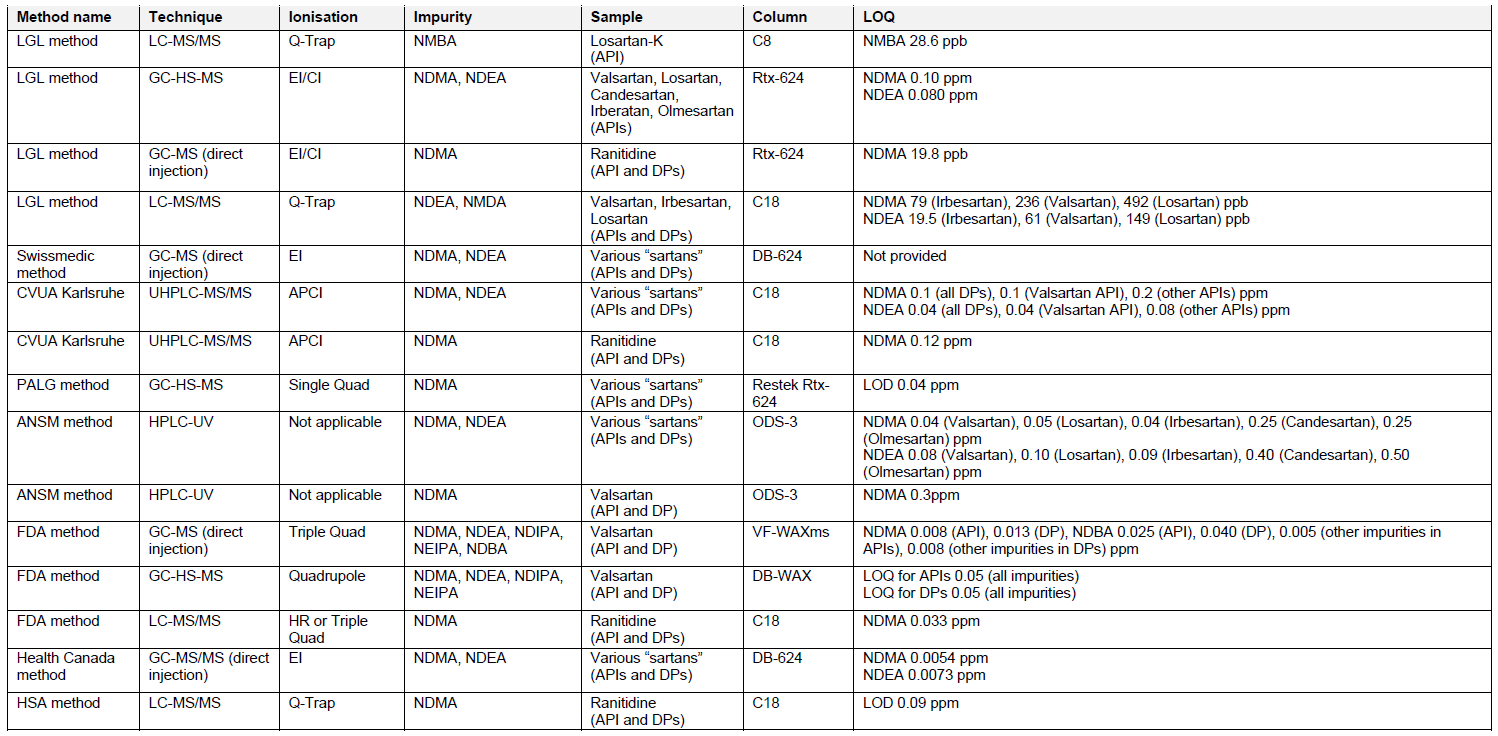
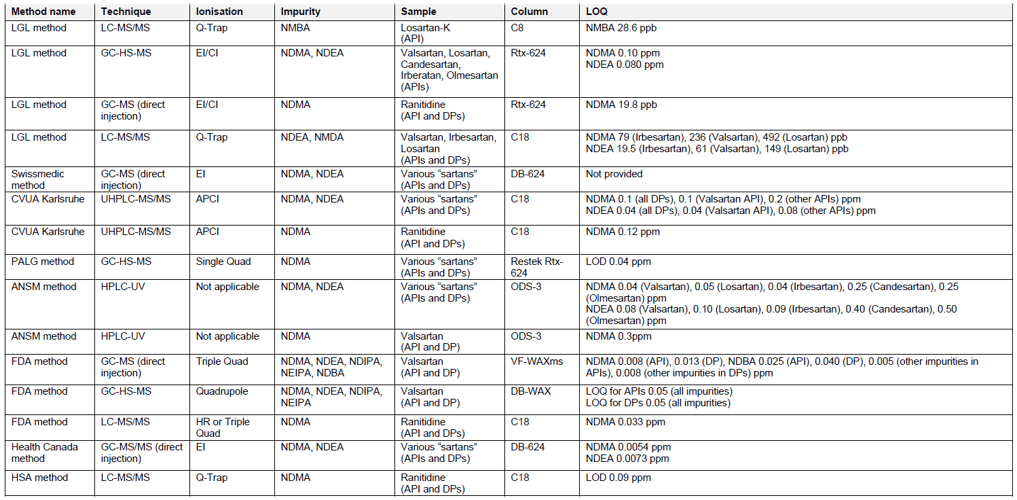
There are several analytical methods which can be utilized for the testing of APIs and finished drug formulations for the presence of nitrosamines, published by the Official Medicines Control Laboratories (OMCLs), FDA, Health Canada, and Singaporean Health Sciences Authority (HSA). The overview of these methods is provided in Table 2. The methods for testing of nitrosamines in “sartans” involve the use of chromatographic techniques (reversed phase liquid chromatography – RP-LC or gas chromatography – GC), combined with mass spectrometry (MS), spectrophotometry (UV) or nitrogen chemiluminescence (NCD). The identification and accurate quantification of impurities using MS often requires use of isotope labelled internal standards. Reference standards of nitrosamines can be obtained in the ready-to-use methanol solutions, which limit the exposure of analysts to potentially carcinogenic substances. Sample material has to be homogenized, extracted and filtered prior to analysis. Nitrosamines are known to be light sensitive, therefore samples should be prepared freshly before testing and the use of amber glassware or other protection against the light is recommended. Some nitrosamines may also degrade at elevated temperatures, e.g. applied in the GC inlet. The suitable sample pre-treatment and use of protective pre-columns should be considered.
Special attention should be given to the specificity of selected analytical methods, considering various sample matrices and the interference of sample compounds with investigated impurities. Analysis of model, spiked solutions, and real product samples with the subsequent evaluation of peak resolution and analyte recovery will increase the reliability of the method. Usually, the simple sample handling workflow, without complex extraction or separation procedures and selection of commonly used techniques results in the development of cost-effective, robust, and reproducible methods, which can be applied in the routine analysis. All methods used for the assessment of nitrosamines in APIs and drug products should be validated as per ICH Q2 guideline.