FDA Approval Process Overview
The Food and Drug Administration (FDA), as part of the United States (US) Department of Health and Human Services, is the regulatory agency responsible for the review, approval, safety oversight, and overall control and supervision of food safety, tobacco products, dietary supplements, prescription and over-the-counter pharmaceutical drugs (medications), vaccines, biopharmaceuticals, blood transfusions, medical devices, electromagnetic radiation emitting devices (ERED), cosmetics, animal foods & feed and veterinary products. The FDA approval process for bringing a new pharmaceutical to market is quite lengthy and can span anywhere from 5-20 years. Broadly speaking, the process begins with preclinical research, filing an investigational new drug (IND) application to initiate clinical trials examining efficacy and safety in humans, and seeking regulatory approval with a ?new drug application (NDA). Read further to learn these steps in detail.
1. Preclinical Testing
New chemical entities (NCEs) are compounds found during drug discovery that look promising for use in treating disease. During this phase of research, the toxicity, pharmacokinetics, and metabolism of the compound are explored. In addition, drug properties such as chemical makeup, stability, and solubility are established.
2. Investigational New Drug Application
After completing preclinical research, a company must submit an Investigational New Drug (IND) application. The IND is a comprehensive document covering not only all knowledge of the compound to date (drug manufacturing characteristics, chemical composition, and behavior) but also detailing the intended plan for human trials. As part of the IND, all data related to preclinical research and testing must be submitted.
3. Clinical Trials
There are 4 phases of clinical trials: Pre-Marketing Phases 1-3 and Post-Market Phase 4.
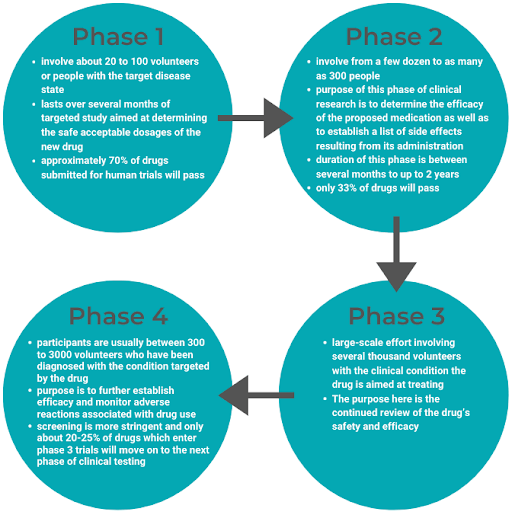
Upon clinical trial completion, all research findings to date are submitted to the FDA in the form of an NDA. Once the FDA receives the NDA, there is a two-month window to determine if the NDA will be accepted for review.
4. NDA Application
FDA review teams scrutinize each NDA submitted. The review teams comprise doctors, pharmacologists, chemists, and other experts to critically evaluate the preclinical and clinical data associated with the drug. The FDA review teams submit evaluation reports with recommendations to the FDA's administrative arm, the administrative body responsible for approving or rejecting the NDA.
5. Drug Labeling
The FDA will also review the labeling and packaging of a potential new drug to ensure appropriate and accurate information is being communicated to both healthcare personnel, as well as regular customers.
6. Facility Inspection
An FDA team visits the site where the drug will be manufactured to inspect the production facilities. This is a routine part of the drug approval process.
7. FDA Drug Approval
After considering the NDA and visiting the production facilities, a review team will decide the fate of the drug. If the team determines that the benefits of the drug outweigh the attendant risks, then they will approve the drug. If not, the FDA will issue a complete response letter denying approval.
How to Get FDA Approval for Your Pharmaceutical Project
The FDA approval process is complex and lengthy with negative financial implications to sponsors if a drug is rejected. Outlined below are 6 compliance tips to improve your chances of gaining FDA approval:
1. Avoid Errors in Documentation
The FDA will present potential drug sponsors with many forms that have to be carefully and correctly filled. An incorrectly filled form can delay the process of drug approval. Avoiding errors in documentation is one of the easiest ways to improve gaining FDA approval.
2. Provide Clear and Measurable Data
It is important when submitting an NDA to the FDA that data from your trials is presented in a clear manner. When a claim of a proposed benefit is made, a reference from clinical research should be provided to back it up. The idea is to provide convincing evidence that a new drug has more advantages than associated risks. Merely claiming a drug is beneficial without providing research-based backing is one of the major causes of denied approval.
3. Focus on Accuracy
Keeping accurate records of all stages of drug development from preclinical testing through clinical trials provides the FDA review team with a comprehensive overview of the drug to date.
4. Ensure Statistical Expertise
Despite a drug’s promise, if data is not properly presented there is a high likelihood that the FDA will not approve it. Consider employing statistical experts to ensure an appropriately sized test group is selected and research data is collected, analyzed, and presented in a relevant and accurate format.
5. Maintain Manufacturing Best Practices
The FDA will routinely inspect manufacturing facilities as part of their approval process. It is vitally important that facilities are always inspection-ready and that good manufacturing practice standards are being followed. This might be the difference between a decision to approve or issue a complete response letter.
6. Communicate Early and Often
Sponsors and researchers involved in the process of developing a new drug are free to ask for clarifications from the FDA at any stage of drug development. The FDA will answer questions regarding the pre-IND application and clinical trial expectations, as well as provide advice on NDAs.
Close communication with the FDA will reduce the need for repeated review cycles and help increase the chances for approval.
At ProPharma, we are dedicated to providing top quality workforce solutions for industries all over the world. We pride ourselves in our ability to create a cordial, long-lasting and valuable employer-employee relationship between the companies we work with and the staff we recruit for them. Contact us today.
TAGS: Investigational New Drug (IND) Regulatory Sciences Labeling