How to Manage the Risk of Elemental Impurities with ICH Q3D: The mission of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) is to ensure safe, effective, and high-quality medicines are developed and distributed. This is accomplished by creating worldwide harmonization. Harmonization achievements in the Quality area include:
- Pivotal milestones such as the conduct of stability studies
- Defining relevant thresholds for impurities testing
- A more flexible approach to pharmaceutical quality based on Good Manufacturing Practice (GMP) risk management
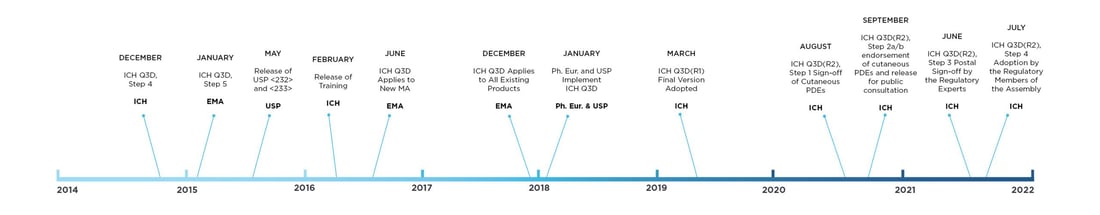
On January 1st, 2018, ICH implemented a guideline for elemental impurities, ICH Q3D, which provides a platform to develop a risk-based strategy to control and limit elemental impurities. As a consequence, this can have a major impact on drug development and more specifically on the quality of your drug.
In every stage of the drug development process impurities can arise. Impurities are substances that are added or formed during the development process and have a non-intentional (potential toxic) effect on the drug. They can be organic molecules, residual solvents, or inorganic compounds. Elemental impurities are commonly called heavy metals and most of them are toxic for humans. Therefore, the levels of elemental impurities must be managed within acceptable limits in order not to harm the patient.
Development of ICH Q3D Guideline
While initially used to control metals like lead, copper, and other heavy metals that constitute a health hazard, the assessment of elemental impurities (EIs) has gained considerable attention in recent years.
ICH Q3D arose out of a need to develop a globally harmonized guideline and is the culmination of several initiatives intended to modernize the control of EIs in pharmaceutical products. The U.S. Pharmacopeia (USP) had been discussing the modernization of its heavy metals monograph for several years prior to the initiation of the Q3D Expert Working Group. After many years of discussion, in 2016 the European Medicines Agency (EMA) implemented a safety and risk-based approach to control residual catalysts in pharmaceutical products approved to be marketed in the EU. The ICH Q3D now replaces these earlier initiatives.

ICH Q3D Guideline
The ICH Q3D Guideline represents a list of 24 elements, classified into four categories according to the toxicity and probability of occurrence.
It promotes a risk-based approach to assess the presence of EIs in drug products. Since the guideline relates to a drug product’s relevant route of administration, a more specific assessment of actual toxicological risk to the patient can be given. A safety-based Permitted Daily Exposure (PDE) has been developed for the oral, inhalation, parenteral, cutaneous and transdermal routes of administration for all 24 elements that are classified based on safety and relative abundance in nature.

ICH Q3D presents major challenges to testing and risk assessments related to meeting current stringent limits for specific elements to assess patient risk. The likelihood that certain elemental impurities are present in a medicinal product should be determined through a valid risk assessment. An important detail is to ensure whether the controls built into the process are acceptable to limit the level of EIs in the medicinal product. The risk assessment should also clarify whether the proposed control strategies are sufficient or if additional control strategies are needed. When the risk assessment indicates that the level of an elemental impurity may exceed the control threshold, additional measures need to be implemented to ensure that the level does not exceed the PDE.
These additional measures can include, but are not limited to:
- Reduction of EIs to levels that do not exceed the control threshold through purification steps or implementing in-process or upstream controls
- Selection of components of improved quality
- Establishment of specification limits for the drug substance, excipient, or drug product
- Selection of an appropriate container closure system
Based on the outcome of the risk assessment, a clear control strategy needs to be provided.
As elemental impurities can be present in every step of the development of your drug product, the control of EIs should be considered across the entire product lifecycle. Whenever changes are implemented, the risk assessment on EIs should be reviewed and updated because every change may have possible impact on the EI content of your drug product. For instance, changes in synthetic routes, excipient suppliers, materials, processes, equipment, container closure systems, or facilities on the original risk assessment should all be evaluated. Also, the regulatory implications of modifications to the risk assessment and control strategy should be considered, and, when needed, it is important that appropriate variations are submitted.
Elemental impurity data for some components may be limited during drug development, which could direct the Sponsor to a particular control strategy (e.g. Sponsor may choose to carry out end product testing as the initial strategy). As additional experience and knowledge are obtained over time, the Sponsor may determine that a change in the calculation option, risk assessment, and/or control strategy may be warranted to ensure the levels of EIs.
Risk Assessments on Elemental Impurities
Since the implementation of the ICH Q3D guideline on EIs, drug product manufacturers are obliged to carry out a thorough risk assessment to identify and control EIs. Since elemental impurities can occur in the drug substance as well as in the final drug product, all potential sources should be considered in the risk assessment. The risk of any elemental impurity occurring in the product at a level > 30% of its PDE should result in a control strategy for that particular EI:

Because of all the implemented changes, the ICH Steering Committee decided that the development of a comprehensive training program and supporting documentation was necessary. They have developed a training program to ensure the proper interpretation and effective utilization by industry and regulators. Ten training modules are provided to assist industry with the implementation and include examples.
Get your knowledge on elemental impurities up to date now!
TAGS: European Medicines Agency (EMA) International Council for Harmonisation (ICH) ICH Q3D Guideline Regulatory Sciences
