In continuation of its series of guidances on electronic submissions, FDA recently released a guidance for receipt dates of electronic submissions which emphasizes one important fact: word choice matters. In the guidance, FDA outlines subtle differences between the receipt and submission dates, as well as what it means to receive or file an application. While it may seem like semantics, understanding the differences between receipt date and submission date is imperative to complying with FDA deadlines set by the Prescription Drug User Fee Act and the Generic Drug User Fee Act.
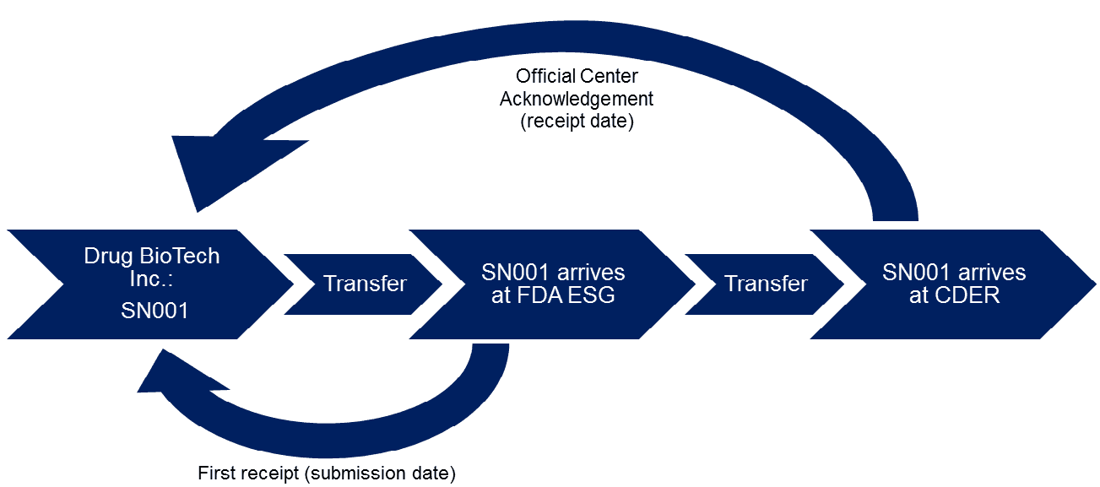
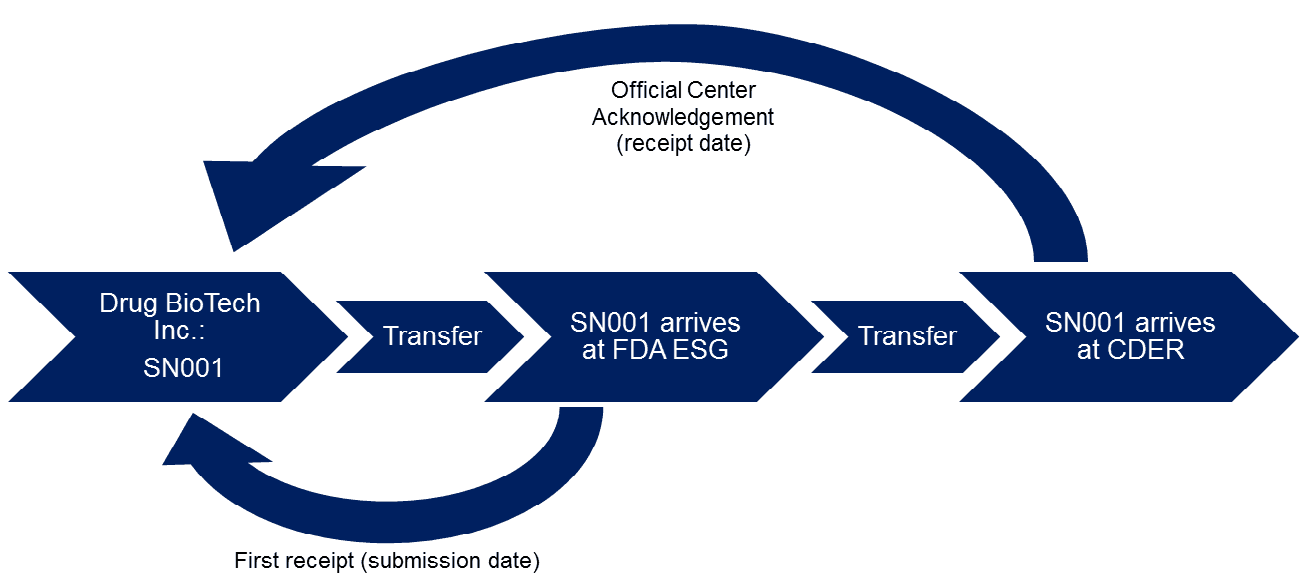
An example of a typical submission process is outlined below for submission of SN001 from Drug BioTech Inc. to the Center for Drug Evaluation and Research (CDER):
The guidance also announced a change in the allowable receipt times for the electronic submission gateway (ESG). Previously, and still cited on their website as of today, any submission received at the appropriate center after 4:30 PM would have a receipt date for the following government business day. Submissions to the ESG can now be made until 11:59 PM without pushing the receipt date to the next government business day. In addition, the ESG is now available to receive submissions from 12:00 AM on Monday to 11:59 PM on Friday (excluding federal holidays and days when the corresponding FDA center is closed).
Deadlines are determined by the receipt date for most FDA submissions, however, in some cases, such as the 180-day exclusivity provision for generics, the submission date holds more significance. While it is true that the submission date and receipt date are usually the same, there are rare occasions in which they may be different, such as if the submission was submitted slightly before midnight (which would be reflected in the submission date) and the receipt is sent slightly after midnight the following day (which would be reflected in the receipt date). FDA also notes that the receipt date should not be confused with FDA’s decision to receive or refuse to receive an ANDA. This decision happens later on and is based on whether the ANDA is deemed to be adequately complete.
ProPharma is a regulatory consulting firm located in the Nation’s Capital dedicated to helping our clients meet their goals through well-developed regulatory strategies. We are experienced in assisting our clients gain FDA approval for their pharmaceutical and medical device products. Should you have any issues in complying with FDA’s new guidance, please do not hesitate to contact us.
If you have any questions or thoughts on this blog post or others, please contact us.
