April 3, 2018
April 3, 2018
On March 20, 2018, the US Food and Drug Administration (FDA) released two new guidance documents to help companies comply with the December 20, 2016 final rule establishing postmarketing safety reporting (PMSR) requirements for combination products with FDA marketing authorization:
These guidance documents were highly anticipated by industry to ensure the appropriate, successful, and timely implementation of the combination product PMSR requirements in advance of the swiftly-approaching compliance date.
Before addressing the impact of these guidance documents, it is necessary to establish why they exist and to whom they apply:
What are combination products?
According to the draft guidance, “a combination product is a product comprised of any combination of a drug, a device and a biological product,” and “each drug, device and biological product included in a combination product is referred to as a constituent part of the combination product.”
Combination products are classified as:
While these definitions are relatively straightforward, some combination products may be difficult to identify. The FDA encourages companies uncertain of the classification of their product to contact the Office of Combination Products (OCP) to request a binding classification or informal classification feedback.
Why does the combination product PMSR final rule exist?
While PMSR regulations for drugs, devices, and biological products are similar and share the same purpose, each set of regulations has distinct reporting standards and timeframes based largely on the characteristics of the product type. Combination products merge product types, blurring these distinctions and warranting an overlap of PMSR requirements to ensure adequate reporting. However, no specific combination product PMSR regulations existed before the final rule, and current application-specific reporting requirements alone yield reporting inconsistencies for combination products.
According to the draft guidance, the final rule “addresses the application of these regulatory requirements to combination products to ensure consistent and complete reporting while avoiding duplication.”
Who is impacted by the combination product PMSR final rule?
According to the draft guidance, the combination product PMSR final rule applies to two types of applicants, including:
The draft guidance also includes combination product PMSR considerations for entities that are not “applicants.” While these entities are not subject to the final rule, they may have postmarketing safety reporting obligations due to their involvement in some aspect of the manufacture or marketing of a combination product.
What do the new guidance documents address?
Practical Application of the Final Rule
The Postmarketing Safety Reporting for Combination Products draft guidance is focused on application of combination product PMSR final rule. The document is lengthy, but its content is highly practical with numerous examples and detailed clarifications.
The draft guidance addresses major provisions of the final rule, including:
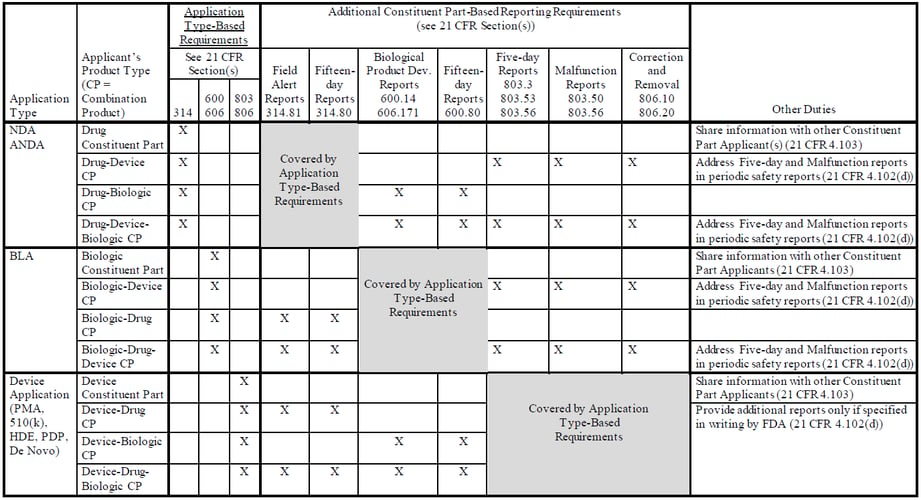
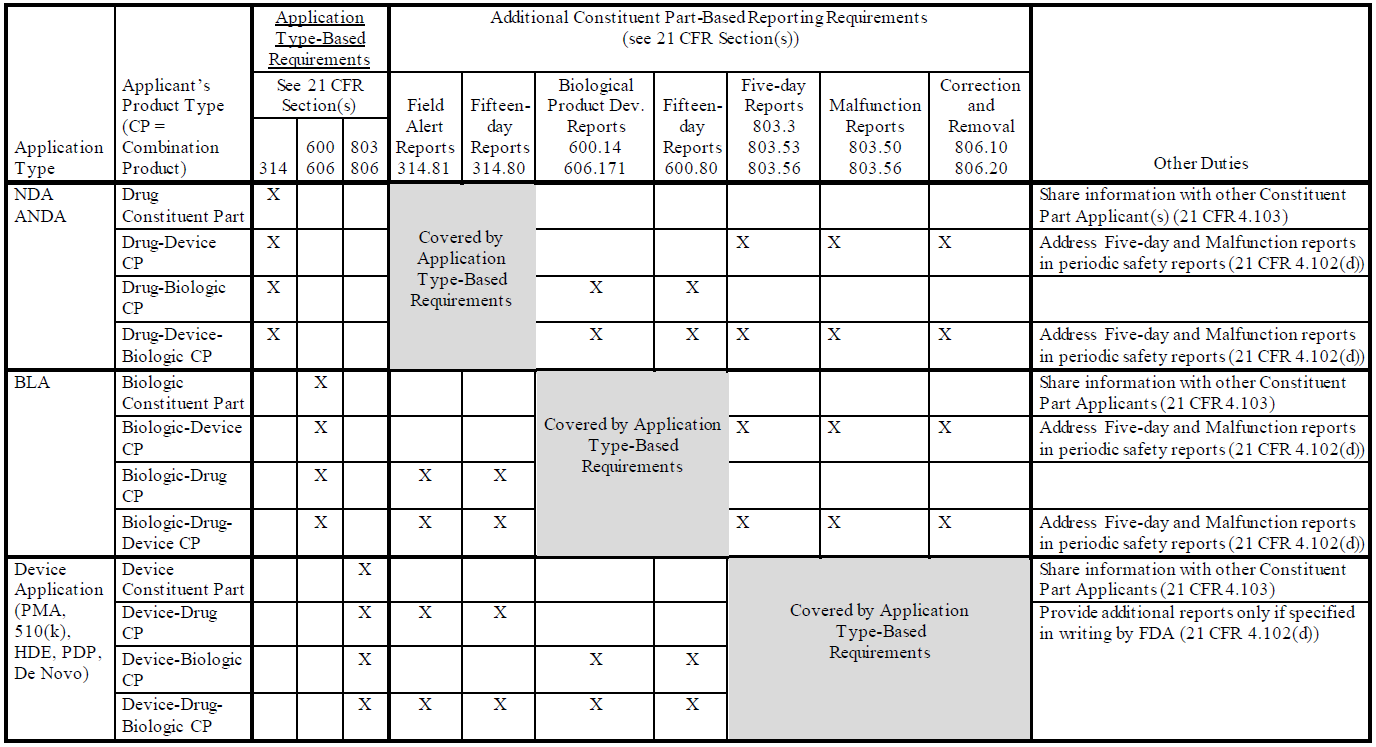
Details regarding the combination product PMSR reporting requirements are summarized in the following table from Appendix 1 of the draft guidance:
Delayed Enforcement Dates
Due to the delay of the draft guidance document and technical specifications for PMSR submissions thereof, many in industry implored the FDA to delay the enforcement of the final rule. The Compliance Policy for Combination Product Postmarketing Safety Reporting guidance addresses the FDA’s compliance policy for the final rule, which includes a delay in the enforcement of specific requirements.
Per the guidance, the following requirements under the final rule will be enforced on the original compliance date of July 19, 2018, as it is believed that significant updates to systems and procedures are not necessary to comply with these provisions:
However, the FDA will delay the enforcement of the following requirements under the final rule:
The enforcement will be delayed until the following dates:
The guidance indicates that the delayed enforcement will “ensure that Combination Product Applicants have sufficient time to update reporting and recordkeeping systems and procedures, including their information technology systems, to comply with these requirements.”
What does this mean?
Industry should be pleased with both the detailed clarification offered by the Postmarketing Safety Reporting for Combination Products draft guidance and the delayed enforcement of select provisions of the final rule outlined in the Compliance Policy for Combination Product Postmarketing Safety Reporting guidance.
However, industry may find it difficult to fully comply with the final rule until the regional technical specification document for combination product ICSR reporting is published and associated updates to DTD and the safety reporting portal are made, at a minimum. Once available, global pharmacovigilance database providers can make necessary database and report configurations, including the addition of structured data fields for constituent parts, and organizations can establish associated processes and data entry conventions.
While implementation of the provisions of the final rule now seems tenable, issues remain.
As with others in industry, ProPharma Pharmacovigilance is currently updating reporting and recordkeeping systems and procedures to comply with the combination product PMSR requirements in advance of the specified compliance dates.
Are you ready?
TAGS: Pharmacovigilance
April 13, 2016
In September 2007, the Prescription Drug User Fee Act (PDUFA IV) was reauthorized and expanded, broadening and strengthening FDA’s drug safety program. As part of PDUFA’s reauthorization, the Agency...
October 26, 2021
The FDA regularly issues new and revised product-specific guidances to facilitate the availability of generic drugs and assist the generic pharmaceutical industry with identifying the most...

May 18, 2022
The FDA’s Office of Clinical Pharmacology within the Office of Translational Sciences released a new draft guidance document that, for the first time, clearly addresses the FDA’s recommendations and...