
Periodic Safety Update Reports (PSURs) play a pivotal role in ensuring the ongoing safety and efficacy of medicinal products once they've entered the market. While initial clinical trials provide essential insights, they often involve limited participant numbers and exclude certain higher-risk subgroups. As a result, the true scope of a drug's safety profile may not fully emerge until it's in widespread use. PSURs address this gap by continuously evaluating real-world data, identifying rare adverse reactions, assessing safety in special populations, and gauging long-term effects. This blog will review the significance of PSURs, their evolution into Periodic Benefit-Risk Evaluation Reports (PBRERs), and the crucial role they play in maintaining public health.
Let’s start with a little bit of background on PSURs and why they are important.
Background: Why Are PSURs Important?
Need for Continuous Evaluation of Safety and Efficacy
During clinical development, the safety and efficacy of an investigational medicinal product (IMP) is generally based on data derived from randomized controlled trials involving a limited number of participants, exposed to the IMP over relatively short treatment periods. When selecting a study population, inclusion/exclusion criteria are applied to ensure the participants enrolled are reflective of the treatment population for a products’ intended indication. Often, higher risk subgroups (i.e., paediatric or elderly patients), patients with significant comorbidities (i.e., renally impaired patients or those with genetic abnormalities), and patients receiving medications that might interact with the IMP, are excluded from trials. Close monitoring of trial participants leads to the detection of common adverse drug reactions (ADRs) in the intended treatment population, however insight as to the type and frequency of rare ADRs, the safety of use in special populations, and long-term treatment data is limited. In addition to spontaneous ADR reporting, aggregate safety information emerging from clinical trials is periodically reviewed through the preparation of Development Safety Update Reports (DSURs). In the post-marketed period, a broader range of patients are treated and ADRs too rare to be detected in clinical trials, and populations at increased risk of experiencing ADRs, may be identified. Additionally, new data emerging from larger populations may change the frequency of ADRs previously detected in trials. Therefore, it is essential that throughout the products’ lifecycle, there is a continued evaluation of new data for evidence of relevant safety and efficacy information that may change the products’ benefit-risk profile.
The evaluation of new data is performed promptly, as significant findings are detected, and periodically to allow a cumulative assessment of the accumulating data. Although most new information will be safety-related, new information about effectiveness, limitations of use, alternative treatments, and many other aspects of the drug’s place in therapy may be pertinent to its benefit-risk assessment.
Evolution of Reporting Mechanisms
The concept of the Periodic Safety Update Reports (PSUR) was first introduced in 1992 and was later harmonised in 1996 by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). This provided a common format for the presentation and evaluation of the safety experience of a medicinal product at defined times post-approval. Safety data reported in the post-marketing setting, and safety data reported in DSURs during clinical development, are included in the evaluation.
The initial primary objective of the PSUR was to provide a comprehensive picture of the safety of approved medicinal products to determine if changes were needed to the Reference Safety Information (RSI). However, people began to realise that the assessment of the risk of a medicinal product is most meaningful when considered in light of its benefits. Subsequently, a new report was proposed, one providing greater emphasis on benefit than the PSUR, particularly when there is a significant change to risks. In such cases an overall explicit evaluation of benefit-risk is required. And so, in 2012 the ICH began creating the guidelines for the newly minted “Periodic Benefit-Risk Evaluation Report” (PBRER).
Standardization and Regulatory Updates
In December 2013, following finalisation of the ICH guideline on “PBRERS”, the European Medicines Agency (EMA) published the updated Module 7 of the Guideline on Good Pharmacovigilance Practices (GVP) Revision 1 (Rev 1), harmonizing the principles of cumulative data presentation, assessment and reporting of PBRER, introducing regional appendices, and instructions for the PBRER assessment process. Rev 1 updates also included instructions for the application and description of the EU reference date (EURD) list following first publication on 01st October 2012. Information included in the EURD list is maintained by the EMA.
Which Products Require PBRERs and How Often Are They Submitted?
The EURD list determines which products require PBRERs and when they must be performed. It is comprised of a list of active substances of medicinal products subject to different marketing authorisations (MAs), together with the corresponding EU reference dates, frequencies for submission of PSURs and related data lock points. The requirement for PSURs for generic, well-established use, homeopathic and traditional herbal medicinal products was waived as per amended Directive 2001/83/EC, however PBRER submissions for they products may be requested by a competent authority in a Member State. Such a request could be based on safety concerns or a lack of information regarding active substance use after authorization.
If required, PBRERs are submitted on 1, 2, 3, 5, 6, 8, 10, 13, 15 or 16 yearly cycles, depending on the product.
PBRER submissions to competent authorities in non-EU markets may be required for a variable number of years following marketing approval, or on an ad hoc basis, depending on the competent authority/region.
For applicable products, Marketing Authorisation Holders (MAHs) are responsible for submitting PBRERs for their own products according to the following timelines:
- within 70 calendar days of the DLP (day 0) for PSURs covering intervals up to 12 months (including intervals of exactly 12 months);
- within 90 calendar days of the DLP (day 0) for PSURs covering intervals in excess of 12 months;
- within 90 calendar days of the DLP for the submission of ad hoc PSURs requested by competent authorities unless otherwise specified.
What is the Purpose of PBRER?
The objective of the modern PBRER is to promote public health and to optimize public safety by providing a comprehensive, concise, and critical evaluation of the benefit-risk balance of a post-marketed medicinal product at defined points in its life cycle. The stages of the evaluation are summarized in Figure 1.
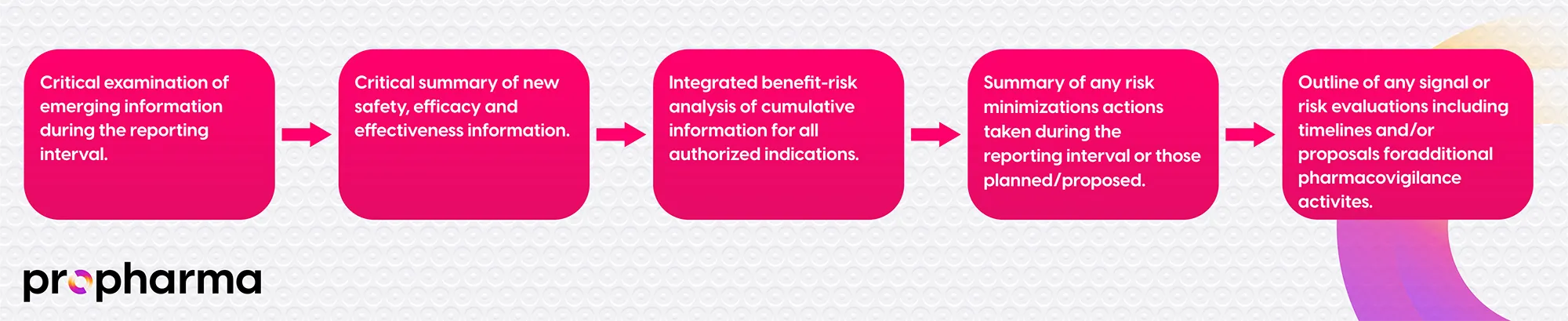
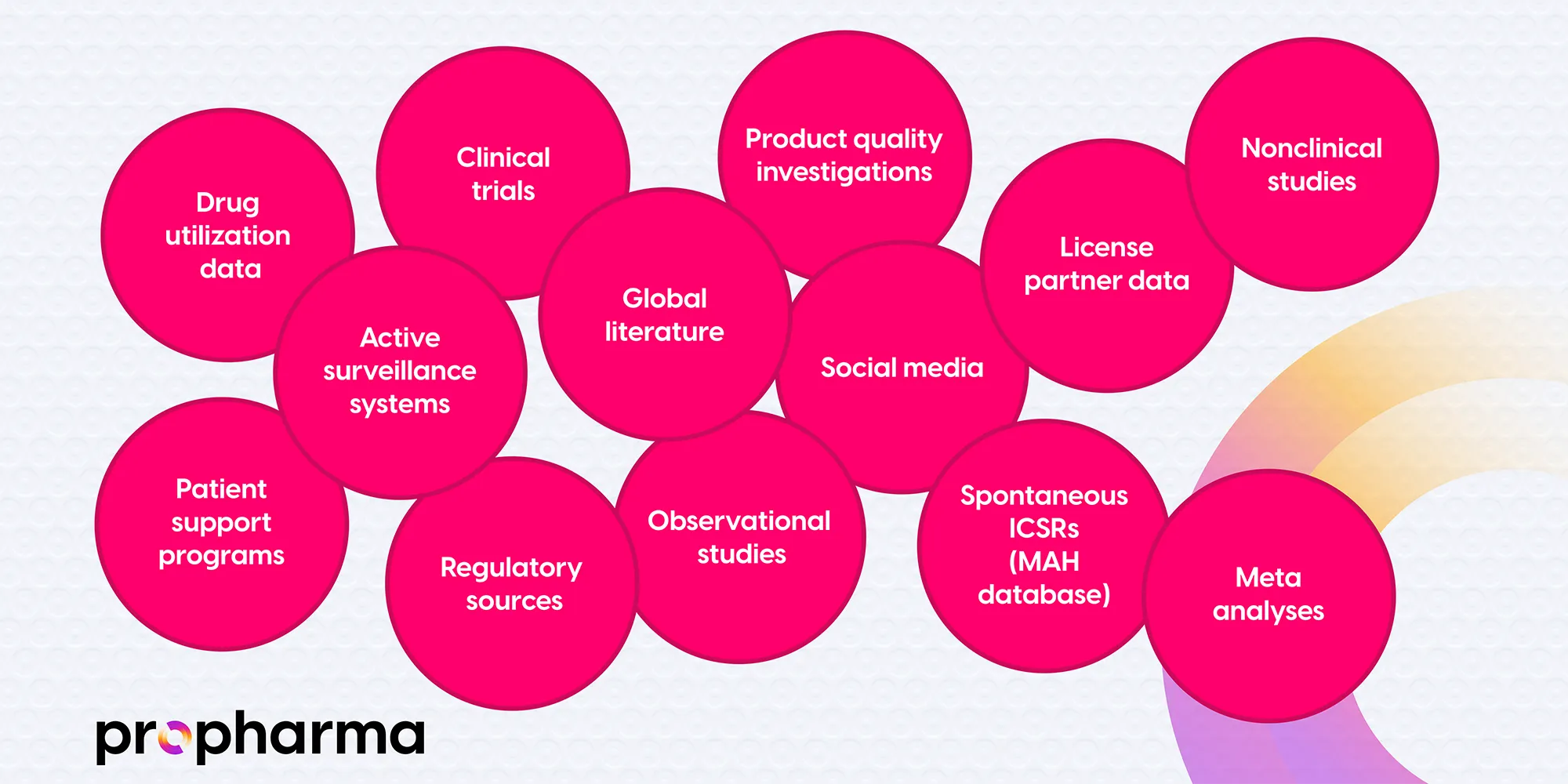
The report includes clinical trial and post-marketed data, an evaluation of important identified and potential risks, and the effectiveness of risk minimization measures performed for them. Further sources of data evaluated in the PBRER are presented in Figure 2.
Let Experts Support Your PSUR / PBRER Submissions
Internationally, the requirement for PBRERs, and the periodicity of submission varies greatly, and it can all get a little confusing for MAHs to stay on top of. At ProPharma, a team of experts in performing periodic reviews of aggregate data have created and maintain a schedule of regulatory and commercial requirements for aggregate reports, delineated by product and geographic region. This tool assists MAHs in maintaining their product licenses by keeping track of their responsibilities regarding PBRER submissions, when they need to be submitted, and to which competent authorities. With a depth of experienced medical and pharmaceutical scientists at hand, ProPharma offer services in the timely preparation, review, and submission of high quality PBRERs.
For more information and to get help with your PBRER submissions, contact us today to speak with an expert.
TAGS: Pharmacovigilance PSUR Periodic Benefit-Risk Evaluation Report (PBRER)


