On March 20, 2018, the US Food and Drug Administration (FDA) released two new guidance documents to help companies comply with the December 20, 2016 final rule establishing postmarketing safety reporting (PMSR) requirements for combination products with FDA marketing authorization:
- Postmarketing Safety Reporting for Combination Products – Draft Guidance for Industry and FDA Staff (March 2018)
- Compliance Policy for Combination Product Postmarketing Safety Reporting – Immediately in Effect Guidance for Industry and Food and Drug Administration Staff (March 2018)
These guidance documents were highly anticipated by industry to ensure the appropriate, successful, and timely implementation of the combination product PMSR requirements in advance of the swiftly-approaching compliance date.
Before addressing the impact of these guidance documents, it is necessary to establish why they exist and to whom they apply:
What are combination products?
According to the draft guidance, “a combination product is a product comprised of any combination of a drug, a device and a biological product,” and “each drug, device and biological product included in a combination product is referred to as a constituent part of the combination product.”
Combination products are classified as:
- Single-entity combination products – Constituent parts are physically, chemically or otherwise combined or produced as a single entity [21 CFR 3.2(e)(1)]
- Examples include syringes, injector pens, and/or applicators prefilled with drug, drug-eluting stents, transdermal patches, and monoclonal antibodies combined with therapeutic drugs
- Co-packaged combination products – Constituent parts are separate entities packaged together in a single package or unit [21 CFR 3.2(e)(2)]
- Examples include metered-dose inhalers co-packaged with a filled drug product cartridge and first-aid kits containing bandages, gauze, antibiotic ointments, and pain relievers
- Cross-labeled combination products – Constituent parts are packaged separately but specifically labeled for use together [21 CFR 3.2(e)(3) or (4)]
- Examples include photosensitizing drug and activating laser/light source and an intrathecal infusion pump intended only for use with an individually-specified drug
While these definitions are relatively straightforward, some combination products may be difficult to identify. The FDA encourages companies uncertain of the classification of their product to contact the Office of Combination Products (OCP) to request a binding classification or informal classification feedback.
Why does the combination product PMSR final rule exist?
While PMSR regulations for drugs, devices, and biological products are similar and share the same purpose, each set of regulations has distinct reporting standards and timeframes based largely on the characteristics of the product type. Combination products merge product types, blurring these distinctions and warranting an overlap of PMSR requirements to ensure adequate reporting. However, no specific combination product PMSR regulations existed before the final rule, and current application-specific reporting requirements alone yield reporting inconsistencies for combination products.
According to the draft guidance, the final rule “addresses the application of these regulatory requirements to combination products to ensure consistent and complete reporting while avoiding duplication.”
Who is impacted by the combination product PMSR final rule?
According to the draft guidance, the combination product PMSR final rule applies to two types of applicants, including:
- Combination Product Applicants – An applicant who holds the only application or all applications for a combination product.
- Constituent Part Applicants – An applicant who holds an application for a constituent part of a combination product, the other constituent part(s) of which is marketed under an application held by a different Constituent Part Applicant.
The draft guidance also includes combination product PMSR considerations for entities that are not “applicants.” While these entities are not subject to the final rule, they may have postmarketing safety reporting obligations due to their involvement in some aspect of the manufacture or marketing of a combination product.
What do the new guidance documents address?
Practical Application of the Final Rule
The Postmarketing Safety Reporting for Combination Products draft guidance is focused on application of combination product PMSR final rule. The document is lengthy, but its content is highly practical with numerous examples and detailed clarifications.
The draft guidance addresses major provisions of the final rule, including:
- Application Type-Based Reporting Requirements – These requirements are based on the application type under which the combination product or constituent part received marketing authorization and apply to both Combination Product Applicants and Constituent Part Applicants.
- Constituent Part-Based Reporting Requirements – These additional requirements are based on the type(s) of constituent parts included in the combination product and apply only to Combination Product Applicants. These constituent part-based requirements include:
- Device constituent parts
- Five-day reporting requirements (21 CFR 803.3, 803.53, and 803.56)
- Malfunction reporting requirements (21 CFR 803.50 and 803.56)
- Correction or removal reporting and recordkeeping requirements for events that do not require a report (21 CFR 806.10 and 806.20)
- Drug constituent parts
- Field alert reporting requirements (21 CFR 314.81)
- Fifteen-day reporting requirements (21 CFR 314.809)
- Biological product constituent parts
- Biological product deviation reporting requirements (21 CFR 600.14 and 227 606.171)
- Fifteen-day reporting requirements (21 CFR 600.80)
- Streamlining Options – These options allow Combination Product Applicants to submit a single report to comply with more than one reporting requirement (e.g., application type-based and constituent part-based requirements), provided the reports can be submitted in the same manner and the combined report satisfies all applicable reporting requirements, including all submission timelines.
- Information Sharing – The final rule requires a Constituent Part Applicant to share with another Constituent Part Applicant information regarding death or serious injury (21 CFR 803.3) or an adverse experience (21 CFR 314.80(a) and 600.80(a)) associated with the combination product within 5 calendar days. Only initial information in its original format needs to be shared. No report or analysis is required.
- Records Retention. The final rule specifies what records Combination Product and Constituent Part Applicants must maintain and how long to maintain them.
- “Constituent Part Applicants must retain PMSR records for the time-periods stipulated in the regulations applicable to its type of constituent part.”
- “Combination Product Applicants must retain records for the longest time-period required for records under all PMSR requirements applicable to the combination product.”
- Device constituent parts
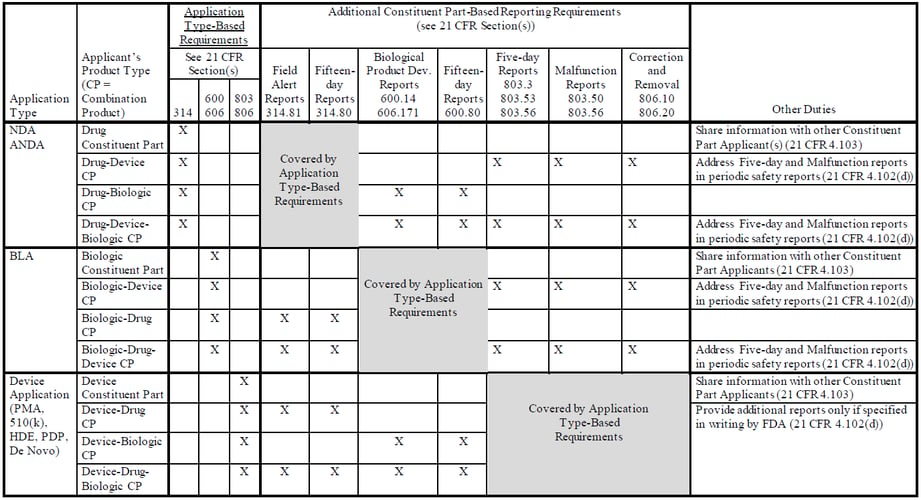
Details regarding the combination product PMSR reporting requirements are summarized in the following table from Appendix 1 of the draft guidance:

Delayed Enforcement Dates
Due to the delay of the draft guidance document and technical specifications for PMSR submissions thereof, many in industry implored the FDA to delay the enforcement of the final rule. The Compliance Policy for Combination Product Postmarketing Safety Reporting guidance addresses the FDA’s compliance policy for the final rule, which includes a delay in the enforcement of specific requirements.
Per the guidance, the following requirements under the final rule will be enforced on the original compliance date of July 19, 2018, as it is believed that significant updates to systems and procedures are not necessary to comply with these provisions:
- Application type-based safety reporting requirements for Combination Product Applicants and Constituent Part Applicants (21 CFR 4.102(a) and (b)),
- Submission process requirements for Constituent Part Applicants (21 CFR 4.104(a))
- Recordkeeping requirements for Constituent Part Applicants (21 CFR 4.105(a)(1))
- Information sharing requirements for Constituent Part Applicants (21 CFR 4.103) and
- Recordkeeping requirements for information shared with Constituent Part Applicants (21 CFR 4.105(a)(2))
However, the FDA will delay the enforcement of the following requirements under the final rule:
- Constituent part-based safety reporting requirements for Combination Product Applicants (21 CFR 4.102(c) and (d))
- Submission process requirements for constituent part-based Individual Case Safety Reports (ICSRs) for Combination Product Applicants (21 CFR 4.104(b))
- Recordkeeping requirements for Combination Product Applicants (21 CFR 4.105(b))
The enforcement will be delayed until the following dates:
- July 31, 2019 for Combination Product Applicants using the FDA Adverse Event Reporting System (FAERS) and Electronic Medical Device Reporting System (eMDR) to report ICSRs
- January 31, 2020 for Combination Product Applicants using the Vaccine Adverse Event Reporting System (VAERS) to report ICSRs
The guidance indicates that the delayed enforcement will “ensure that Combination Product Applicants have sufficient time to update reporting and recordkeeping systems and procedures, including their information technology systems, to comply with these requirements.”
What does this mean?
Industry should be pleased with both the detailed clarification offered by the Postmarketing Safety Reporting for Combination Products draft guidance and the delayed enforcement of select provisions of the final rule outlined in the Compliance Policy for Combination Product Postmarketing Safety Reporting guidance.
However, industry may find it difficult to fully comply with the final rule until the regional technical specification document for combination product ICSR reporting is published and associated updates to DTD and the safety reporting portal are made, at a minimum. Once available, global pharmacovigilance database providers can make necessary database and report configurations, including the addition of structured data fields for constituent parts, and organizations can establish associated processes and data entry conventions.
While implementation of the provisions of the final rule now seems tenable, issues remain.
- The combination product PMSR requirements are specific to the FDA. A report formatted with FDA-dictated regional specifications may also require global submission. Will manual adjustments need to be made to the report or will global pharmacovigilance database providers restrict the transmitted data based on region? Will efforts be made to harmonize product-specific approvals and reporting requirements globally?
- Departments responsible for handling adverse events and product complaints are often separate. Greater collaboration and communication between these departments will be required from intake to submission to ensure complete compliance with combination products PMSR requirements, specifically for those combination products with device constituent parts.
- It may not be possible to assign causality to specific constituent parts in current pharmacovigilance databases. Future database and report configurations may be required.
- Constituent Part Applicants for the constituent parts of a single combination product receive, exchange and store PMSR data separately. Are unique safety data exchange agreements necessary to resolve issues regarding the exchange of information and data ownership between Constituent Part Applicants?
As with others in industry, ProPharma Pharmacovigilance is currently updating reporting and recordkeeping systems and procedures to comply with the combination product PMSR requirements in advance of the specified compliance dates.
Are you ready?
TAGS: Pharmacovigilance Combination Products Postmarketing Safety Report (PMSR)
