Roadmap for Successful IVDR Transition: The compliance dates for the In Vitro Diagnostics Regulation (IVDR) will become effective on May 26, 2022. To help you with the IVDD to IVDR transition, we've created this comprehensive guide with everything you need to know.
If you're interested in expert guidance to help with the IVDR transition, contact us to learn about our services.
Contact UsWhat is IVDR?
IVDR is the new regulation for in vitro diagnostics in the European market. Currently, diagnostic products distributed in the European Union must be certified and comply with the In Vitro Diagnostic Directive (IVDD). Both IVDD and IVDR focus on quality and safety, but the IVDR strengthens these expectations for both notified bodies and manufacturers across the entire product lifecycle, from early development to distribution to eventual discontinuation. To achieve this goal, the IVDR focuses more on the supplier management, as well as the quality management system in general.
Impact of IVDR on Quality Management Systems
Quality management systems are going to be greatly impacted. Updates to QMS will need to be executed prior to the transition. To determine how rigorous your quality management system needs to be, the IVD classification needs to first be determined, as the risk-based approach employed in the IVDR is like that used by the FDA and for medical devices in the EU under the Medical Device Regulations (MDR). One of the biggest changes under the IVDR is that many more products will now require a notified body certification before they can be distributed.
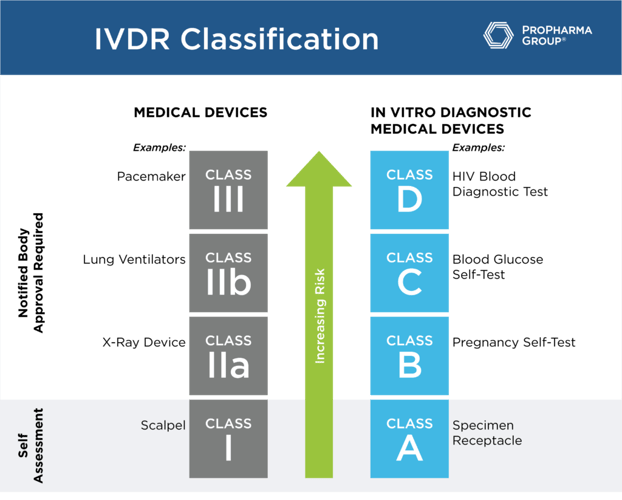
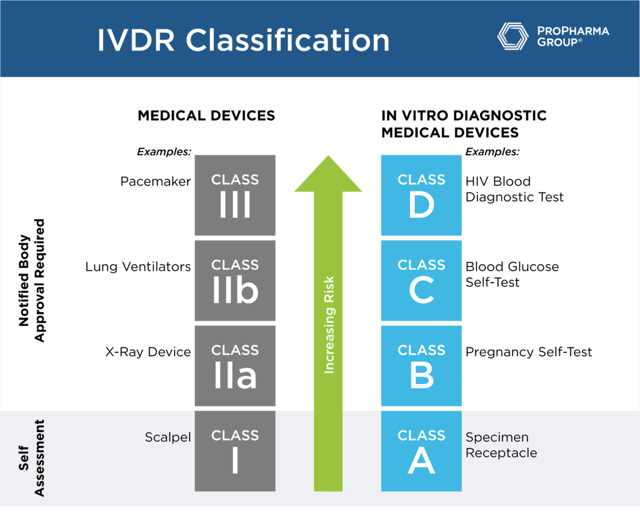
While the MDR uses Class I through Class III based to designate risk classification, under the IVDR, the classifications are A, B, C and D (see table below). For class A, a self-assessment by the manufacturer that has a certified quality system will suffice. These are the only products that won't need to undergo notified body review of the technical files because they are the lowest risk. For class B, C and D, notified body review of technical files, in addition to having a fully compliant quality system, is a requirement.
For the QMS transition of the EU IVDR, the following applies. If you already have an IVDD certification for your IVD, you will have an essential requirement. This has been switched over to execute a general safety and performance requirement (GSPR). To comply with the regulation, the items on this IVDR Self-Assessment checklist will be essential. The biggest updates can be found on the topics of management responsibility, performance evaluation, CAPA monitoring, and measuring. The IVDR will ensure manufacturers are in line with ISO 13485:2016. Some of the biggest changes are in risk management. The ISO 14971 standard Medical Devices – Application Of Risk Management To Medical Devices has continued to evolve quickly. While the EU has sometimes lagged the FDA, manufacturers will need to understand the changes in the 2019 version of the standard when compared to the 2000 and 2007 version and ensure their products are compliant to this new standard. Topics such as product realization or design control have been updated.
Roadmap for Successful IVDR Transition: Next Steps for a New or Existing Product With IVDD
Do you currently have a product in distribution under the IVDD? Are you launching a new product for the European Union market? How do you transition to the IVDR?.
A priority is to find a notified body if you don't have one. Next, you need to determine the classification of your products and perform a gap analysis of your quality systems to evaluate your readiness for ISO 13485 certification. This will include all your processes, your work instructions, your forms and your records. What is your quality system compliance to IVDR like, and how can you ensure compliance moving forward? If you already have a notified body, verify they are transitioning to IVDR.
A next step is to determine the number of gaps based on the IVD requirements. Research those findings and address them as CAPAs to ensure there is a plan for closing all gaps through both corrective actions and preventive actions. Finally, all technical documentation must be updated from the IVDD to IVDR requirements that will then allow you to schedule your inspection through one of your notified bodies. If you're a new company and you're not in the European Union market yet, you will need to start with your quality management system assessment to ensure that it is compliant with ISO 13485:2016.
ISO 13485 – Quality Management for Medical Devices
For regulatory purposes, ISO 13485 is the standard by which notified bodies will evaluate your systems for certification. The standard identifies the requirements specific for medical device companies including IVDs. It ensures consistency of design, production, and installation sufficient to support appropriate quality standards for products distributed in the EU. It starts with your quality management system at a high level, meaning both quality manuals, as well as your quality policy and quality objectives. These high-level documents are followed by management responsibilities. For compliance with the standard, a company must also ensure the job descriptions and training for employees based on their skill sets and job function are in place
Finally, product realization is the critical aspect of your design control for your IVD. Delivering an acceptable product to the marketplace must not only ensure proper manufacturing controls but must also include supplier management and deliver the expected product based on customer requirements and user needs. With more stringent requirements regarding manufacturing, importing and distribution, companies need to make sure that contract manufacturers are identified, a quality agreement is in place, and audits are performed appropriately
Technical Documentation Requirements for IVDR
Understanding your classification is important in terms of the regulatory burden that you're going to face in this transition to being compliant with IVDR. The conformity assessment by a notified body is required for class B, C, and D devices before placing a device into the market. Companies are no longer allowed to self-certify these types of products. So, in order to meet the regulation requirements, all the technical documentation must be in place. Also, if you haven't renewed the certificate within the past year, you're going to have a significant amount of work to do to prepare the submission to the notified body for all but class A devices (general lab use devices and some instruments).
To put the technical documentation together, the first question a manufacturer should ask is, what's the intended use of the product? This is a requirement for the compliance with IVDR. Changes to the intended use can have an impact on other files with other regulatory agencies, for example your registrations with the FDA, with Australia and with the Chinese regulatory bodies. So, although you are often adding more detail to be compliant with IVDR, you may have to manage how this additional detail could affect your registration with other regulatory agencies and how these other regulatory agencies want to address the interconnectivity or the impact they may have in their country
Software Analysis and Classification for IVDR
Under the new IVDR regulation, there's an emphasis on software and its impact to classification. Types of questions that you will need to ask yourself are: Does the software just run the instrument? Is the software an algorithm that will interpret the results from an assay or provides a medical decision?
The answers impact the classification of your device and the technical documentation you need to provide. This also increases the burden on manufacturers providing a post market surveillance plan. Previously, this wasn't a significant or substantive component of technical documentation, although manufacturers within their quality systems regulations had these outlined. There is a new emphasis on these post market surveillance plans that need to be included with technical documentation, as well as periodic safety update reports. If your device evolves over time, notified bodies will expect updated documents that reflect those changes.
Other types of questions that will need to be answered are: Does the software run the overall functioning of the instrument? Does the software simply load or move samples within the instrument? Does the software operate independent of the device? How is the software involved in interpreting the result? Is it interpreting a measurement and providing a result or giving a clinical output? These are all important characteristics to understand how the software is operating because they will impact the classification, and therefore the documentation you must provide in your submission.
The next point of consideration is the performance evaluation plan. The performance evaluation plan is composed of both your analytical studies and your clinical studies. This is tied to the specifications and the requirements of the device. You would need to know for example, how was the assay cut off determined at 0.7 and what does that mean? Who was it determined that the assay needed to have a certain cutoff?
This and many other questions need to be asked and answered as you evaluate the specifications and requirements of the device. Finally, is the post market surveillance plan robust? Does it cover all the appropriate facets (software, instrumentation, and complaints) of the device? Are the complaints that may affect the performance of the device taken into consideration? How are you planning to compile the complaints, reports or changes that have occurred in terms of possible suppliers or third-party manufacturers that you are using? These should all be part of the post market plan for it to be adequate to comply with the new IVD regulations.
The performance evaluation plan includes the items that the notified body will want to see when going into documentation review. Is the clinical performance study plan up to date? Does it have rationale for how the clinical performance of the device was validated? The performance evaluation plan should include the analytical study reports, the clinical study reports, and the numerous post market reports that must be considered in your quality management system. These should be supportive and alleviate any concerns that a notified body may have when reviewing the technical documentation.
Pitfalls and Considerations
Here is an overview of key considerations:
- You must determine how software affects your classification of your instruments. If you have a plate that has software that is only moving a plate from one place to another, or that's allowing the instrument to aliquot certain volumes of your reagent into wells, you likely have a class A device. For these products, manufacturers don't need to make a submission to a notified body to put the device on the market, because they are not required to submit technical documentation. Moving from a class A to a class B or higher risk device will lead to more regulatory burden. That is, change request assessments by a notified body will be required when you make a change to an instrument or a platform. This change notification can greatly complicate and slow the speed of change, especially when third party manufacturers are involved. The time to introduce the change is not only based on the notified body review, but also agreeing on the timeline for implementation with the third party.
- Once you are clear on the classification, you have to generate the documentation and ensure that the quality management system is adequate to comply with the regulations. The compliance of your quality systems to the regulations will be the central focus of any audit.
Project and Organization Management Questions Related to IVDR
What does it mean to have strong project management for a successful implementation?
Clear understanding of the objectives, scope, deliverables, timelines, ownership, and "definition of done." Given the 150+ pages of IVDR regulation, those elements along with identification of the integration points will facilitate implementation of IVDR activities as there are impacts, responsibilities, and required collaboration across multiple functions.
Can you pursue remediation to IVDR without a strong project management structure?
Sure. But, you will risk not being compliant to a critical piece of regulation, lacking transparency on project progress towards IVDR compliance, missing critical IVDR timelines, potential duplication of efforts across multiple stakeholders, and missing critical issues that could have been identified as a result of assessing compliance to the regulation. Hence, having some form of project management structure is important for implementation of IVDR requirements and successfully maintaining your presence in the EU market.
IVDR Checklist and Deadlines
Our experts have put together a short IVDR Self-Assessment checklist to help you assess your implementation of the IVDR regulation using core, basic questions as the barometer of your progress. As mentioned in Part II of this blog series, it is important that you have the intended purpose and classification of your product defined first as well as sign-off from your core team and senior leadership sponsors on those items.
Why is that important? Because this provides the foundation for how you complete your IVDR implementation. For example, if you have a class B versus a Class D product, the volume of tasks that will need to be completed are much greater for one class versus the other – resulting in more deliverables, greater stakeholder involvement, and greater accountability.
As of the writing of this piece, there are four notified bodies certified for IVDR: BSI, DEKRA, TÜV SÜD, and TÜV Rheinland. It is important to understand your notified bodies' plans to become certified for IVDR, associated timelines, and your contingency plan should they not get certified for IVDR. If you do not have a notified body, you will need to determine the best one for your organizational, regulatory, and geographic needs.
One often missed preparation for the IVDR certification is a mock inspection. This will allow you to assess regulatory readiness via documentation, processes, technology, and governance. This will also challenge you to understand whether your processes are truly fit for purpose and meet the spirit of the regulation. Finally, your process leads can assess their ability to effectively speak to the new regulation or processes they oversee.
A Case Study
An In Vitro Diagnostics client had executive and senior leadership support within their PMO and functions but did not have enough subject matter experts to deliver the program due to the many daily work priorities competing with IVDR readiness. The IVDR program also struggled with retaining their external skilled experts, putting in jeopardy their ability to meet the May 2022 deadline.
We quickly enhanced their program governance structure, identified and staffed experts to close out the skill gaps, confirmed understanding of devices classifications, project management deliverables (e.g., charter, project plan), and definition of done. We updated or newly developed required IVDR documentation and confirmed process owners. We helped them ensure that project resource attrition was minimized, deliverables were appropriate to the scope of the device's portfolio, and content will be ready and available to share with notified bodies for the certification process per the deadlines.
Sustainability After IVDR Certification
Once you have achieved certification with IVDR, you must now sustain the process. Sustainability means proactively ensuring that your processes are fully optimized and in alignment with any newly produced amendments and guidances, have ongoing monitoring and continuous improvement of the processes, and deliver ongoing training/communications to support the evolving needs. This is critical to ensure that you are fully aligned with current IVDR expectations and your devices meet the current standards.
As a reminder, the new IVDR is effect as of May 26, 2022 impacting all IVD companies. ProPharma Group is actively helping multiple diagnostic clients navigate their way through the change to regulations in the European Union. If you need support, please contact our team.
We help clients ensure that they are compliant with the current directive as well as transitioning through MDR/IVDR so that they can continue to market compliant products in the European Union. Contact us for support.
TAGS: In Vitro Diagnostics (IVD) In Vitro Diagnostic Regulation (IVDR) ISO 13485 Medical Device Regulations (MDR) Medical Devices