Phase-Appropriate Development and Application of Quality Systems in the Drug Development Process: The Parenteral Drug Association (PDA) recently published a revised version of Technical Report No. 56: Application of Phase-Appropriate Quality System and cGMP to the Development of Therapeutic Protein Drug Substance (API or Biological Active Substance). The FDA-harmonized guidance is welcome news to organizations involved along the drug commercialization continuum illustrated below1.
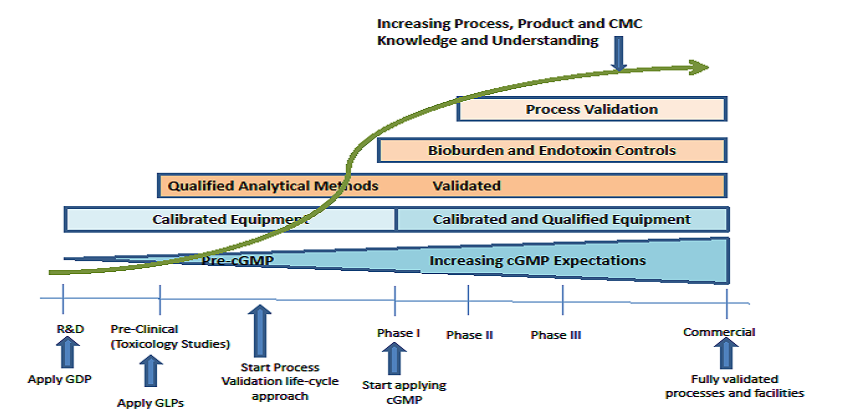
The guidance provides clarity for the industry, allowing organizations to focus on implementing phase-appropriate recommendations and requirements, depending on their current development state and the evolution of their quality system. Equally valuable, it serves as a longer term “road-map,” providing vision for planning and implementation of increasing regulatory compliance complexity as progress is made through the commercialization process.
Phase-Appropriate Development and Application of Quality Systems in the Drug Development Process (con't): For example, a R&D start-up that decides to conduct toxicology studies knows that Good Documentation Practices (GDP) and Good Laboratory Practices (GLP) are requirements prior to initiating the studies. Towards that end, they can implement GDP/GLP soon enough to support the toxicology testing phase. The same start-up can avoid headaches and costs by mistakenly implementing cGMP before they are really needed. Also note that organizations that fail to implement the minimum phase appropriate requirements run the risk of having any data generated during the R&D phase be considered suspect by regulatory reviewers, e.g., data generated using uncalibrated equipment or unqualified analytical methods could be considered to lack an appropriate level of integrity.
For those organizations on the left side of the process, the message is also clear that their development work is critical since it serves as the basis for the more stringent and complex manufacturing control strategies, CMC, process validation, method validation, and other requirements necessary to compliantly drive a product to market. The regulatory expectation is that the knowledge gained during development phases continues to grow in terms of manufacturing process understanding and control.
While large established drug developers may have the resources necessary to plug a new drug into their existing commercialization systems and mechanisms, the same cannot always be said for biotech start-ups, small research outfits, and academic research labs.
ProPharma Group possesses the experience and expertise necessary in assisting organizations to understand and implement the phase-appropriate quality system requirements along the entire continuum. Contact us to discuss how we can assist your organization make wise choices and maximize your drug development efforts.
1 Illustration imitated from PDA Technical Report No. 56: Application of Phase-Appropriate Quality System and cGMP to the Development of Therapeutic Protein Drug Substance (API or Biological Active Substance) – (Revised 2016).
Interested in learning more? Contact us today to find out how we can help with your global regulatory needs.

