
Compiling and submitting a New Drug Application (NDA) submission is a complicated and intensive activity. Once you have submitted your application to FDA, you may be curious about what can you expect to hear and when. Below are some of the common questions we receive from Sponsors about what to expect during the NDA review process.
When Will I know That my NDA Has Been Accepted for Filing?
Once you submit your NDA, FDA's review team will determine if it is complete. If it is not complete, FDA can refuse to file the submission. If the NDA is complete, the review team has varying timelines for review, depending on the type of application, to decide whether the drug will receive FDA approval. Per the Prescription Drug User Fee Act of 2022 (PDUFA VII), FDA's must meet the following review timelines:
- Standard New Molecular Entity (NME) NDA – within ten months of the 60-day filing date
- Priority NME NDA– within six months of the 60-day filing date
- Standard non-NME original NDA – within ten months of receipt
- Priority non-NME original NDA – within six months of receipt
You will know within 60 days of submitting your NDA to FDA whether it has been accepted for filing (or conversely, if FDA refuses to file your application due to lack of information or studies).
FDA Day 74 Letter
If your application is accepted for filing (60 days after submission), you will receive a FDA Day 74 letter (74 days from receipt of the original NDA submission), which contains a planned NDA review timeline. FDA's review timeline will vary based on several factors, such as whether other similar drugs already exist or whether your drug is intended to treat an unmet medical need. The planned review timeline included in the 74-Day letter will include timelines for:
- The PDUFA user fee goal date
- Postmarketing requirement/commitments
- Proposed labeling
- Advisory Committee (AC) (if anticipated)
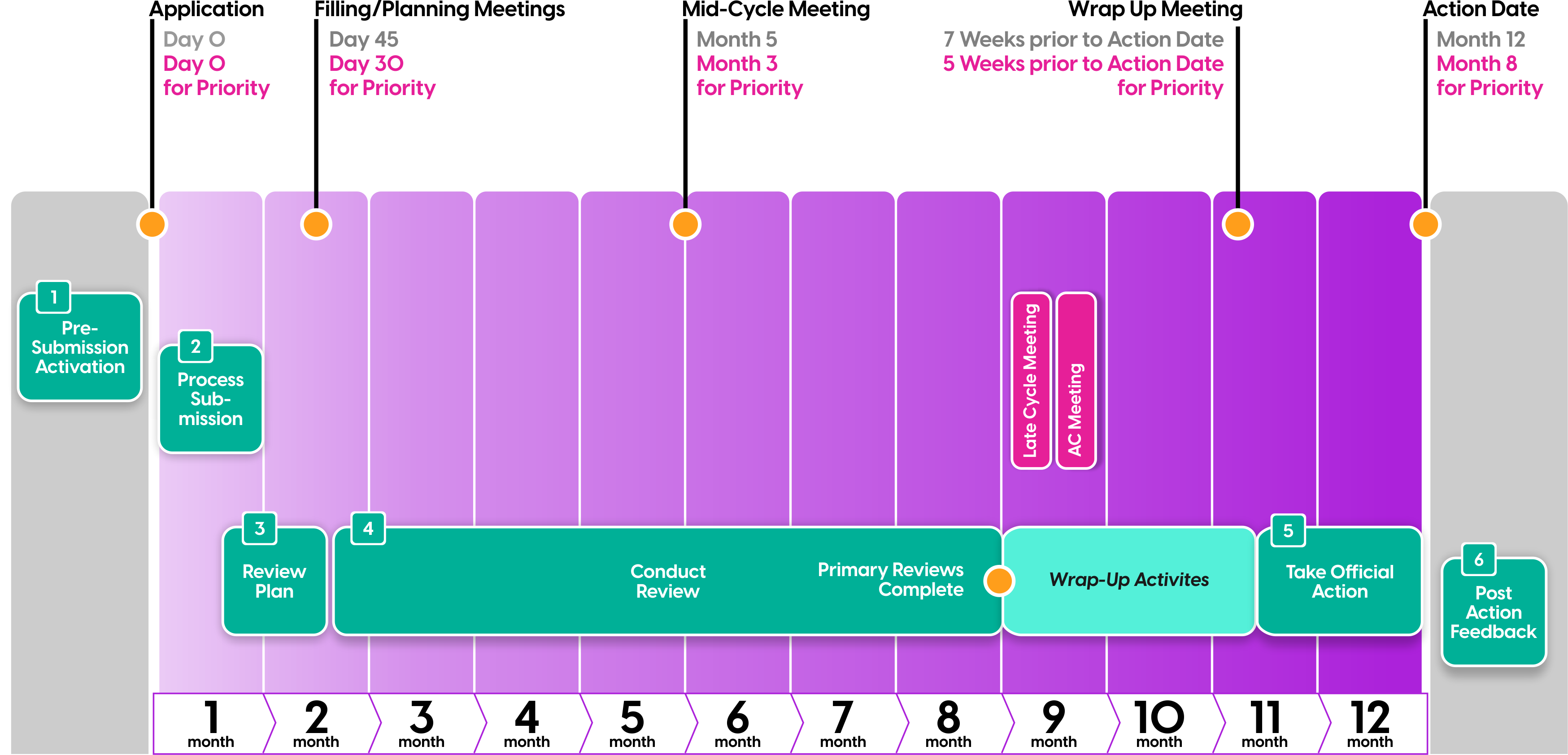
The FDA has established internal review timelines as described in their guidance document, which included the timeframes for FDA internal milestone meetings (filing, planning, mid-cycle, team, and wrap-up meetings). FDA will inform you of any necessary information requests or status updates following the milestone meetings or at other times throughout the review process.
Furthermore, the FDA 74-Day letter also confirms standard versus priority review, and identifies any preliminary deficiencies in your application. Sponsors must act quickly to resolve the deficiencies noted in the FDA 74-day letter during the NDA review process., so it's advisable to seek outside assistance from an experienced regulatory affairs consultant.
When Can I Receive Information Requests from the FDA?
Mid-Cycle Meeting
During the review cycle, FDA will hold an internal mid-cycle meeting by month five for standard review (month three for priority reviews). During this meeting, FDA’s objectives include:
- Present status and key findings of all reviews, consults, and inspections
- Confirm the decision that was made regarding the need for an Advisory Committee meeting
- Identify any issues that could preclude an approval action
- Begin high-level discussion of labeling (e.g., are major claims supported) and need for post-marketing requirements (PMRs) and/or post-marketing commitments (PMCs)
- Determine if a REMS is needed (if not already determined) and, if so, the goals and the elements of the REMS
- Revise the review plan and interim timelines (if needed)
- Solicit feedback from the signatory authority and other discipline directors
- For expedited reviews, discuss possible early target date(s) for completion of reviews and action
Although FDA can request information from the Sponsor at any time during their review, Sponsors can expect FDA communication including a number of information requests within two weeks before and/or after the mid-cycle meeting. Sponsors must act quickly to resolve any information requests provided by FDA during the review period.
FDA Inspection
You should also be prepared for an inspection of the facility or facilities where the drug will be manufactured. FDA’s goal is to complete all GCP, GLP, and GMP inspections for applications within six months of the date of original receipt for priority applications and within ten months of the date of original receipt for standard applications. The inspection may result in additional requests at the end of the review cycle.
How Many Information Requests Can We Expect to Receive from FDA During the NDA Review Process?
The answer is that it varies, and there really is no predicting how eventful your review will be. Throughout the course of the Agency’s review, there are likely to be issues that come up, but the number and significance of these requests will vary from application to application. Hopefully you took the time to ensure that all components of your NDA were accurate and complete, and you reviewed any previous correspondence with the Agency to make sure all issues were addressed. However, FDA may request clarification regarding where information resides in your NDA, clarification on information you already provided, or they may request additional analyses.
Regardless of how many information requests you receive during the NDA review process, it is important that you make sure you understand what FDA is asking for and that you respond with information that is clear, comprehensive, and transparent. This will help FDA’s reviewers understand your response and minimize the need for additional follow-up questions or requests. It is the Sponsor’s responsibility to respond to these requests as soon as possible, as timely and complete responses help ensure that you don’t inadvertently push out your action date.
When Does Label Negotiation Occur During the NDA Review Process?
Label negotiation is a critical step that takes place at the very end of the review, in many cases in the last month of review. Tensions often run high during this period since many parties have an interest in the final wording of the label. You should have developed strong, data-driven statements about your product prior to submission, but now you must to be prepared to defend those statements. Timing and responsiveness is critical at this stage, so it is essential that all important members of your team are available to review versions of labels and participate in calls on very short notice.
NDA Submission and Review Timeline
The NDA review process is long and complex, so it is critical to ensure you’re aware of what to expect during each step so you have plenty of time to prepare. The FDA has provided the following NDA timeline to help visualize the entire process.
ProPharma: Experts in Compiling Successful FDA Submissions
Whether you are compiling an NDA for submission to FDA or responding to requests during FDA’s review of your application, it is important to remember that an NDA is the most important and most complex series of documents that you will need to assemble during your product’s lifecycle. An accurate and complete NDA submission is critical to ensuring a streamlined FDA review and maximizing the likelihood of product approval from FDA.
ProPharma - The World's Leading Regulatory Consultancy
TAGS: New Drug Application Agency Alerts General Regulatory Regulatory Sciences


