
Strategic Guidance for Expanding Clinical Trials Across EU Member States with CTIS
Expanding a clinical trial to additional Member States within the European Union (EU) Clinical Trial Regulation (CTR) framework can provide numerous advantages, including:
- Enhanced patient recruitment
- Improved data diversity
- Regulatory alignment across multiple jurisdictions
However, Sponsors must navigate procedural complexities within the Clinical Trials Information System (CTIS) to ensure seamless execution.
Why Expand an Existing Clinical Trial to Additional Member States?
Adding EU Member States to an ongoing clinical trial is a strategic decision that can significantly enhance trial execution, regulatory outcomes, and market preparedness. Key reasons that support expanding an existing trial to additional Member States include:
- Boost Patient Recruitment & Accelerate Enrollment Timelines: If a trial is struggling to meet recruitment targets in existing locations, expanding to additional Member States can open access to new patient populations. Increased enrollment ensures adherence to study timelines and regulatory deadlines, reducing delays in data collection and trial completion.
- Increase Data Diversity & Improve Scientific Validity: Adding Member States allows Sponsors to gather more ethnically, genetically, and environmentally diverse data, enhancing the external validity of study findings. This diversity strengthens the overall quality of the dataset and improves regulatory acceptance across varied populations.
- Align with Market Access Strategies: Expanding trials helps Sponsors generate country-specific data that can support future product authorizations. Regulatory bodies often prefer localized clinical evidence when assessing the efficacy and safety of therapies.
- Address Emerging Regulatory & Ethical Requirements: Some clinical trials may need additional Member States to align with evolving ethical considerations or to meet specific post-approval safety monitoring demands. This can be essential in cases where real-world evidence (RWE) is required for further market expansion.
- Enhance Trial Feasibility & Reduce Risks of Site Underperformance: If certain existing sites are underperforming in recruitment or data collection, adding new Member States allows Sponsors to diversify investigator networks, reducing reliance on limited-site performance.
Navigating Key Regulatory Considerations
Sponsors must ensure alignment of the regulatory documentation required across the different Member States to maintain consistency with prior approvals and prevent unnecessary queries. Differences in country-specific requirements, such as translations for Informed Consent Forms (ICFs), can affect submission timelines and require strategic planning. Additionally, Ethics Committee (EC) variations play a crucial role—some Member States impose rigid compliance expectations, and in specific cases (e.g., France), RFIs on ICFs cannot be addressed, potentially leading to outright application rejection if documentation fails to meet local standards.
Submission timing is another critical factor to consider. Once an amendment is filed, additional modifications cannot be introduced until approval is granted. Sponsors should consider bundling submissions whenever possible to optimize efficiency, minimize administrative burden, and avoid prolonged review cycles.
Understanding the Approval Timeline
The approval process follows a structured timeline:
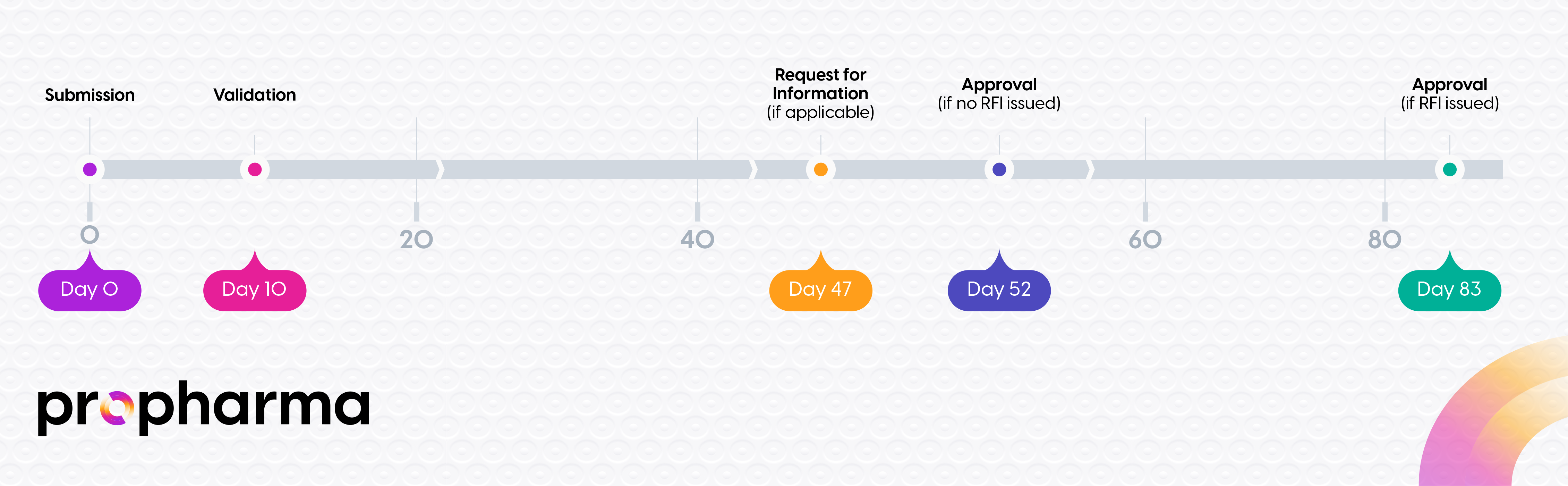
Proactive RFI Management: Strategies to Accelerate Clinical Trial Approval in the EU
RFIs often arise when regulatory authorities in newly added Member States seek further clarification on Part 2 documentation. Common areas of inquiry include ICFs, site-specific compliance, safety monitoring protocols, and study design justifications. Since RFIs can extend approval timelines, Sponsors should proactively prepare responses to prevent unnecessary delays. Establishing structured internal response protocols, maintaining a centralized regulatory database, and leveraging regulatory expertise ensures responses are precise and aligned with CTIS requirements.
Swift communication with regulators and preemptive document reviews can significantly reduce approval risks. Additionally, conducting post-submission analyses of RFIs received enables Sponsors to refine submission strategies, improving efficiency for future trial expansions.
Best Practices for Expanding Clinical Trials Across Member States
Successfully expanding a clinical trial to additional EU Member States under the Clinical Trials Regulation (CTR) requires more than simply replicating the initial submission. Each country may have unique expectations and regulatory nuances, especially when it comes to Part 2 documentation. This can lead to potential Requests for Information (RFIs), which can ultimately delay approvals. By learning from common challenges and proactively refining their approach, Sponsors can streamline the expansion process and avoid costly setbacks.
The following best practices highlight key strategies to anticipate issues, strengthen submissions, and accelerate approval timelines when adding new Member States:
- Anticipate RFIs & Prepare Thorough Documentation: Sponsors should proactively address expected concerns by ensuring submission materials are comprehensive, well-structured, and country-specific.
- Ensure Ethical & Regulatory Compliance Across Member States: Inconsistencies in Part 2 submissions often lead to scrutiny—establishing rigorous internal quality control measures ensures compliance with each jurisdiction’s standards.
- Strategic Site Selection: Expanding trials successfully depends on choosing sites with strong recruitment potential, investigator expertise, and a clear understanding of regulatory obligations.
- Streamline Submission Timing & Amendment Strategies: Filing amendments in bundled submissions rather than piecemeal requests ensures smoother approval pathways, preventing regulatory bottlenecks.
By integrating these lessons learned, Sponsors can optimize the process of adding Member States, ensuring efficient approvals while minimizing regulatory complications.
Seamless Expansion of Clinical Trials with ProPharma
Expanding your clinical trial to additional Member States within the EU Clinical Trial Regulation (CTR) framework can be a complex and demanding process. Partnering with our team of expert regulatory affairs consultants enables you to gain the expertise needed to navigate regulatory requirements efficiently. We provide comprehensive support at every stage, ensuring seamless submissions and minimizing approval delays.
From initial submission to final approval, we handle all regulatory intricacies to help Sponsors successfully expand their trials across Europe. Our services include document preparation, ensuring all regulatory submissions comply with CTIS guidelines, minimizing errors, and reducing risks of delays. We engage directly with regulatory agencies to address inquiries and resolve potential obstacles. Additionally, our expertise extends beyond compiling and submitting regulatory submissions. We proactively assist in responding to Requests for Information (RFIs) and managing regulatory follow-ups to keep your trial on track.
By partnering with ProPharma, Sponsors gain access to deep regulatory expertise, ensuring compliance with evolving European clinical trial requirements. Our dedicated team of regulatory consultants stay ahead of industry standards, safeguarding submissions against unnecessary delays or rejections. Whether navigating the CTIS or managing multi-state trial expansions, we provide the strategic support needed to streamline regulatory approvals and optimize clinical trial success.
TAGS: European Union (EU) Europe General Regulatory Clinical Trials Information System (CTIS) Regulatory Sciences Clinical Trials

