
In a recent poll conducted by ProPharma Group, the question “What is your biggest GMP auditing challenge?” was posed to Quality professionals in the drug manufacturing industry. The following graph represents the percent response to each of the provided answers:
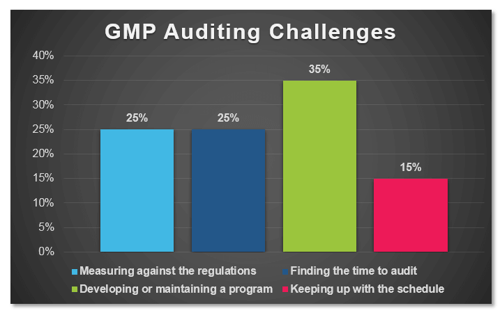
When reflecting on the narrow results of this survey, I realized that it really was not surprising that the top responses “Developing or maintaining a program”, “Measuring against the regulations”, and “Finding the time to audit” did not have much separation as a challenge for Quality professionals.
Developing or Maintaining a Program
It is reasonable that “Developing or maintaining a program” was selected as the leading concern. It can be difficult for firms that already have their own time constraints and quality program obligations to tend to the management or creation of a dynamic audit program. In addition, companies must schedule audits of their suppliers based on the supplier’s schedule and availability, which almost certainly leads to an “end of the year” crunch to schedule audits within the calendar year.
So where can you begin? While there is no way to qualify and manage suppliers without investing significant resources, there are some processes that firms can implement to help manage a compliant GMP auditing program. For example:
- A risk assessment should be performed on all outsourced materials and services to determine the level of risk that failures could have on the quality, safety, and efficacy of the final product. The risk assessment should also evaluate the criticality of the material and the integrity of the supply chain (e.g., reliability of supply, availability of alternate suppliers, etc.).
- Define qualification process for the different levels of supplier risk. For example, suppliers of low-risk, non-critical materials would have less stringent supplier criteria and qualification processes than that of suppliers of high-risk, critical materials. On-site audits should always be performed for suppliers of higher-risk and critical materials / services.
- Perform confirmation testing of materials and components to evaluate if a sample a) meets the defined specifications and b) the certificate of analysis provided with the sample is confirmed through independent testing verification. The level of testing is typically dependent on the assessment of risk (e.g., confirm identification testing only for low-risk materials, full CoA confirmation testing for high-risk materials).
- Implement a Quality Agreement with all suppliers to create mutual understanding of the quality & regulatory requirements relevant to the material service supplied. To monitor routine compliance, it is recommended that the Quality Agreement specify communication requirements for proposed changes and non-conformances that impact the purchased material.
- The performance of critical, high-risk material should be confirmed on a routine basis by performing a quality review and confirmation laboratory testing of the critical quality attributes.
Measuring Against the Regulations
For the respondents who indicated their greatest GMP audit challenge was “Measuring against the regulations”, it is understandable that companies want to benchmark and gauge their quality and compliance programs against what the regulatory agencies expect and what is standard industry practice. After all, the FDA and similar international regulatory agencies specify that the contracting sponsor organization is ultimately responsible for the quality of purchased material and that the contract facilities are considered an extension of the manufacturer’s own facility.
It can be a challenge to measure against regulations when evaluating the compliance of a quality program for suppliers, especially since the FDA regulations are technology-neutral and moreove were designed not to be prescriptive. In addition, there are multiple regulatory requirements and industry accepted guidance covering quality system expectations for all the materials in the supply chain. For example:
- In drug manufacturing, API’s are governed under ICH Q7 guidance
- Excipients are governed under IPEC and/or USP guidance
- Raw materials packaging and processing aids are governed under ISO guidance
Adding to the array of regulatory and industry guidance, many drug manufacturers qualify their critical suppliers by performing on-site audits and document findings with checklists that often limit findings as binary data (i.e., the observed system was either satisfactory or unsatisfactory) without providing detailed, actionable, risk-based conclusions. Without detailed knowledge of the supplier’s quality and manufacturing systems, how can a manufacturer proactively address gaps and areas of concern?
To untangle the vagueness of the regulatory requirements, the multitude of guidance documents that cover the drug manufacturing supply chain, and the lack of audit specificity with the checklist system of reporting findings, we typically recommend that manufacturers implement a systems based audit approach like the FDA six-system inspection model presented in the FDA Compliance Program Guidance Manual (CPGM) for Drug Manufacturing Inspections #7356.002. This manual describes that drug manufacturing can be classified into sets of operations and related activities. The systems examined during these inspections include those related to quality, materials, production, facilities & equipment, packaging & labeling, and laboratory controls. Because overarching systems are thoroughly inspected, one or many product types can be evaluated efficiently within the same context.
Finding the Time to Audit
With the increasing regulatory pressure and expectation that manufacturers qualify their suppliers, it is no wonder that “Finding the time to audit” was another top response to the poll. While we can empathize with the fact that limited resources are required to perform more and more responsibilities, can a manufacturer truly afford not to qualify their suppliers? After all, your suppliers’ problems are your problems.
Navigating the challenges discussed above by increasing the robustness of your GMP auditing program and implementing a risk-based supplier qualification program, your firm can be assured of control over outsourced materials and services. It can also increase the efficiency and utilization of your staff and possibly turn the biggest GMP Audit challenge into a manageable process.
TAGS: Compliance Quality & Compliance Good Manufacturing Practice (GMP)


