
The FDA's Breakthrough Devices Program is intended to expedite the development of innovative technologies for patients with life-threatening or irreversibly debilitating diseases or conditions. The FDA aims to do this by providing medical device and device-led combination product sponsors opportunities for early and regular interaction during the device development process, as well as priority review1 for premarket submissions. The recently updated guidance reinforces FDA's commitment to engage with sponsors to help foster timely innovation that addresses public health problems and disparities.
Breakthrough Device Designation Criteria
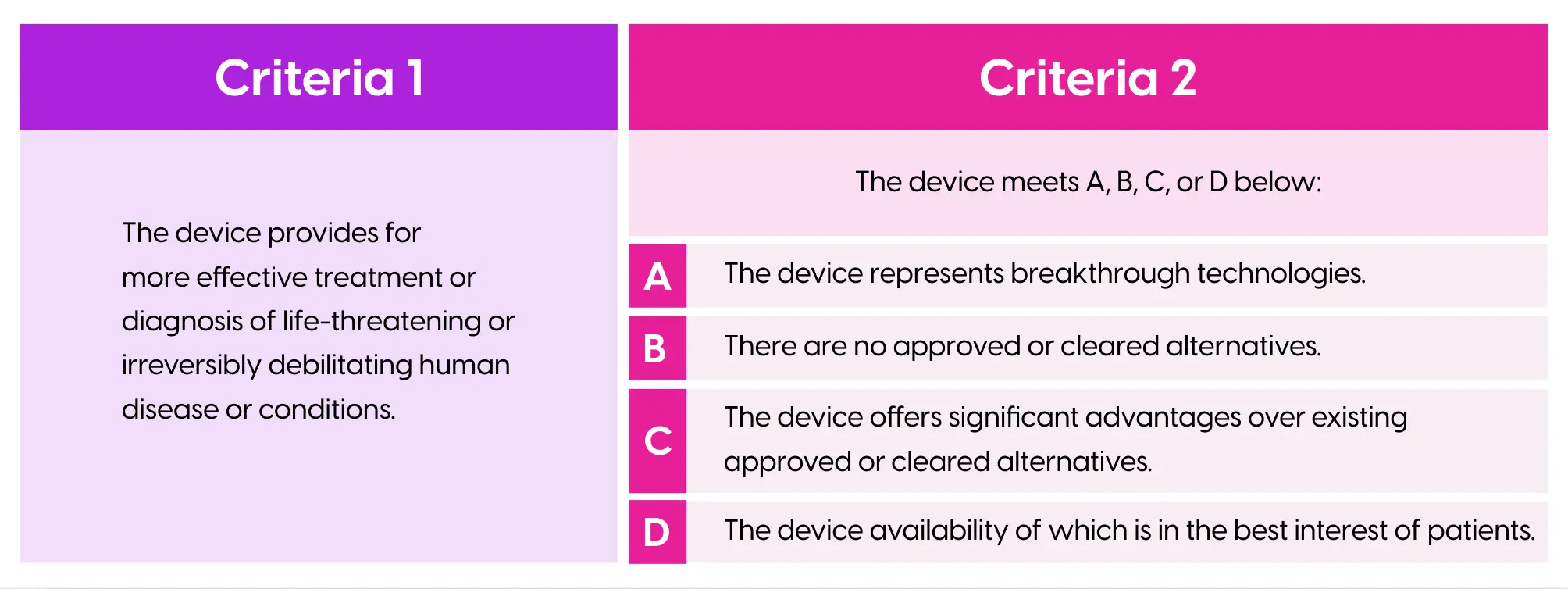
The FDA determines eligibility for the Breakthrough Device Program2 based on two designation criteria, and both criteria must be met in order to receive Breakthrough Device Designation. Figure 1 outlines the designation criteria used in the FDA's assessment for breakthrough device requests. Of note, complete data to support meeting Criteria 1 are not required for entry into the program.
Mechanics of the Breakthrough Device Program
To enter the program, Sponsors must submit a Breakthrough Device Designation Request. This request is a type of Q-submission and should clearly demonstrate how the device meets the program criteria. If the Breakthrough Device Designation is granted, manufacturers are provided opportunities to interact with FDA experts through several different program options, such as Sprint interactions, to efficiently address topics as they arise during the development and premarket submission review phases. These interactions can help sponsors receive and address feedback from the FDA in a timely way.
Unpacking the Updates to the Breakthrough Devices Program Guidance
Updates to the Designation Criteria
Most of the updates to the Breakthrough Device Program guidance reflect the CDRH's effort to promote and advance health equity and provide clarity on the designation criteria.3
Determining if a device meets the first criterion, which involves providing more effective treatment or diagnosis for life-threatening or irreversibly debilitating human diseases or conditions, may not always be straightforward. The FDA has thus clarified that a breakthrough determination is based on the comprehensive assessment of available information, including the device's function, potential for technical and clinical success, potential for clinically meaningful impact, and its potential benefits and risks. The amount and type of evidence needed is based on the intended use of the device, its technology and features, and the available standard of care alternatives.
Regarding health equity, the guidance acknowledges the existence of diverse forms of health care disparities that impact the overall quality of life and health outcomes for all patients. The FDA states that technologies and device features that provide more effective treatment or diagnosis in populations that exhibit health and health care disparities may be considered for the Breakthrough Devices Program when assessing eligibility based on the first designation criteria. Health and health care disparities include differences in treatment outcomes related to race, ethnicity, sex, age, disability and other factors as well as the challenges in recognizing and addressing the reasons behind these differences. Device features and technologies aimed to address differences that arise from social factors, phenotypic variations, pathophysiology, and response to treatment can be reasonably expected to be more effective. The FDA would also consider device features and technologies tailored to the unmet needs of patients with life-threatening or irreversibly debilitating rare diseases under the designation criteria.
Similarly, accessibility to quality healthcare contributes to health and heath care disparities. The FDA defines accessibility as an individual or group's capacity to benefit from a medical device or procedure. Often, underserved populations face barriers preventing them from receiving medical care or diagnosis. Therefore, FDA intends to consider device features and technologies that have the potential to offer a clinically meaningful impact through improved accessibility when assessing the designation criteria. Such features may include adaptability for users, ease of use by diverse populations, and suitability for various settings. Enhancing device accessibility could make it more effective and may lead to better adherence to prescribed medical regimens, particularly among patients who have limited alternative options.
Additional Minor Guidance Updates
There are a few additional minor updates to the final guidance that are summarized below:
- The program may be available for certain non-addictive medical products to treat pain or addiction, consistent with the SUPPORT Act. One of the provisions of the SUPPORT Act is to emphasize development of drugs (and devices) to treat pain that are not addictive particularly for use in chronic pain over time.
- The Designation Request should clearly indicate the proposed indication for use for which designation is being sought.
- When marketing authorization is received, the FDA plans to publicly disclose the Breakthrough Designation status for the device.
How can you find out more about the Breakthrough Device Program?
The FDA plans to hold a webinar on Tuesday, November 14, 2023 to discuss the updated guidance. In addition, if you want to understand how to leverage the interactive nature of the Breakthrough Devices Program to meet FDA's rigorous standards for safety and effectiveness for your device or device-led combination product, ProPharma is here to help put your team on the right track early in your program development. Contact us for support.

