
On April 26, 2023, the European Commission proposed a new package of pharmaceutical legislation1 to revise many of the currently applicable Regulations.
Background
This revision is considered by many as a once-in-a-generation change to the EU's core pharmaceutical legislation, and the proposed legislative updates will have far reaching consequences. Some commonly discussed impacts of the new legislation include:
- Shortened automatic marketing exclusivity periods, and requirements to extend the exclusivity period
- Regulatory sandbox to support innovation and development of new regulatory approaches
- Reduction of review and approval timelines
- The absence of an Environmental Risk Assessment (ERA) as a ground for refusal of the Marketing Authorisation Application (MAA)
- Tighter coordination, monitoring, and management of medicinal product shortages
However, it has been almost a full calendar year since the proposal was released. Where do we stand now?
EU Process for New Legislation
First, let's review the process of New Legislation in the EU from ‘Proposal’ to ‘Application’.
 Step 0:
Step 0:
European Commission puts forward a draft proposal for consultation (minimum 12 weeks) with relevant stake holders - COMPLETE
Step 1:
European Commission puts forward a ‘New’ Pharmaceutical legislation package to European Parliament - COMPLETE
 Step 2:
Step 2:
The European Parliament are expected to review the proposal, possibly amend it, and then vote on whether to adopt it.
However, Step 2 has multiple sub-steps. See the Illustration below generated a in briefing document (June 2023) for the group leading European Parliaments Health Committee (ENVI) who’s Rapporteurs from Germany and Denmark lead the review.
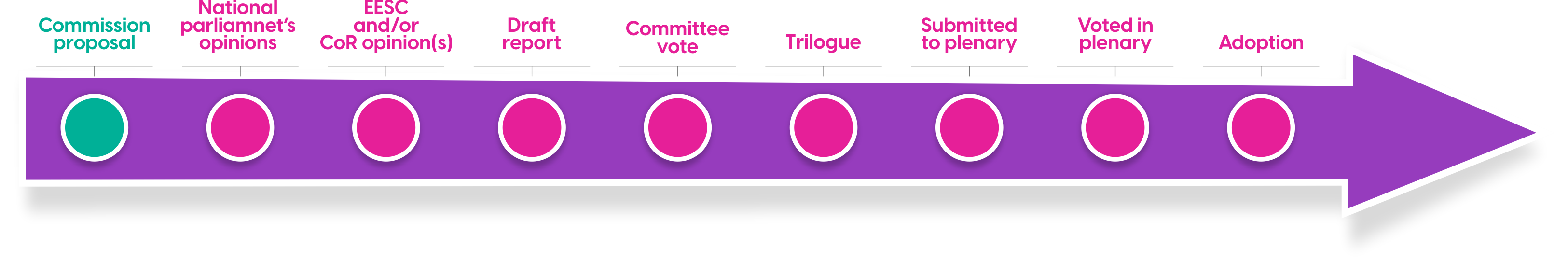
In an ENVI meeting in September 2023, strengths and weaknesses of the European Commission’s Impact Assessment for the new legislation were discussed and broadly determined to be ‘adequate’ though member states expressed various concerns over elements of the new pharmaceutical legislation. It is understood that comments received from ENVI are diametrically opposed on certain key topics including exclusivity periods and regulatory sandboxes.
Optimally, views will converge over Q1 2024, allowing members to present amendments in time for the penultimate plenary session in April. This timing is critical given that European elections will be held in June 2024. If the April time frame is met, the mandate for subsequent negotiations voting and adoption will be put in place. Otherwise, these steps are unlikely to begin until October 2024.
 Step 3:
Step 3:
After adoption timelines are fixed and once adopted, Pharmaceutical Legislative Acts enter into force 20 days after they are published in the Official Journal of the EU. The new Regulation will apply 18 months after publication, the new Directive also applies 18 months, but in this instance, the timeline includes the Directive transposition into local/national law.
Impact of New Legislation and Industry Concerns
The impact on the standard exclusivity period and the introduction of complex criteria for the recovery of lost exclusivity is a major concern for industry. Indeed, it has been reported that EU parliament may modify initial proposals for standard baseline data protection from six to seven and a half years. Furthermore, parliament’s proposal also includes one additional year if the product meets an unmet medical need, bringing it to a total of three 3 years marketing protection, rather than the current two years.
This shift realigns total exclusivity to a maximum 11 and a half years, opposed to the previously proposed 12 years. Orphan standard exclusivity is unchanged at nine years but can be extended to 11 years if the product fulfils an unmet medical need. This is up from a maximum of ten years in the Commission’s original proposal. Proposed changes, such as stricter compliance for ERAs and management of shortages, perhaps represent an incremental development, though complaints of ‘more stick than carrot’ are common from industry.
At first glance impact of the proposed changes to the European Medicines Agency (EMA) structure including the transformation of the Paediatric Committee (PDCO), Committee for Orphan Medicinal Products (COMP), and Committee for Advanced Therapies (CAT) into EMA working parties (Pharmacovigilance Risk Assessment Committee (PRAC) and the Committee on Products for Human Use (CHMP) will remain distinct committees) may not be completely obvious. Conceivably, there is potential for reduced transparency in decision-making within a single entity. However, the actual impact is difficult to predict, and more details may be forthcoming to help in this review. Moving forward it will be wise to review the legislative updates against ongoing development plans and assumptions on a case-by-case basis and ensure integration into future plans.
ProPharma: EU Regulatory Affairs Experts
Drafting new legislation and its implementation can be a slow process in the EU, as the recent EU CTR and the new Pharmaceutical Legislation illustrate. This apparent placid pace can make it difficult for companies to plan and decide when or how to factor changes into their businesses. This is especially true with the added complexity of transitional periods.
In addition, drug development will also be impacted by other (relatively) new legislations on companion diagnostics (Medical Device Regulation (MDR)/ In Vitro Diagnostics Regulation (IVDR)) and health technology assessment (Health Technology Assessment Regulation – HTAR).
Without regular monitoring, review, and a holistic view on legislative developments, it is all too easy for missteps to occur, potentially having a negative impact your product’s development. Want to learn how to avoid any potentially tragic situations from happening to your product? We can help. Contact us today to discuss any concerns you might have and learn how our team of regulatory affairs consultants can help ensure successful regulatory outcomes for your product.
References
TAGS: European Union (EU) Regulatory Sciences EU Pharmaceutical Legislation Reform

