It comes as no surprise to any pharmaceutical or biotech company that planning the clinical development of CAR-T cells is an extremely challenging endeavor:
- high efficacy is expected in each targeted indication. The low safety profile and the high cost of CAR-T cells can only be balanced by an efficacy well above the corresponding standard of care;
- clinical studies to demonstrate a positive benefit-risk ratio include a very limited number of patients (maybe as low as a hundred), who often present diverse baseline demographic/disease characteristics, so as much information as possible should be obtained from each enrolled patient;
- the competence among pharma and biotech companies is very high, with multiple products under development (which could even limit recruitment feasibility) and payers screening which are the most cost-effective therapies to be accessed by the patients with the most urgent needs.
This blog will briefly review the publicly available regulatory documents pertaining to CAR-T cells already approved, in order to better understand the expectations when designing the clinical trials for new products (i.e.: new CARs for different antigens) or new indications.
Most of the challenges, lessons learned and guidance discussed here could be applied to other gene therapy products that are currently under development for the treatment of multiple oncology indications, including off-the-shelf allogenic CAR-T and CAR-NK (natural killer) cells, Supra-CARs, engineered TCR (T Cell Receptor) cells and TILs (Tumor-Infiltrating Lymphocytes) therapy.
Which Are the Currently Approved CAR-T Cell Products?
As of 20th August 2022, 6 different CAR-T cells products have been approved both in US and Europe:
- Abecma® (idecabtagene vicleucel)
- Breyanzi® (lisocabtagene maraleucel)
- Carvykti® (ciltacabtagene autoleucel)
- Kymriah® (tisagenlecleucel)
- Tecartus® (brexucabtagene autoleucel) and
- Yescarta® (axicabtagene ciloleucel)
All of the CAR-T cell products approved to date are autologous products, consisting of the patient's own lymphocytes that have been genetically engineered to raise an immune response against cancer cells. These CAR-T cells have been designed against the CD19 antigen targeting different types of lymphomas and leukemias (Breyanzi®, Kymriah®, Tecartus® and Yescarta®) or the B-cell maturation antigen (BCMA), targeting multiple myeloma (Abecma® or Carvykti®).
Kymriah® and Yescarta®, the undisputed pioneers, were already approved in 2017 (US) and 2018 (EU) but have been granted several new indications since then. And there could be many more CAR-T cells products to come. The development pipeline for such products is presently very active, with multiple clinical trials in different oncology indications, CARs targeting new antigens, allogenic CAR-T and CAR-NK cells, as well as engineered TCR cells or TILs, among others.
How Are CAR-T Cells Used in the Clinical Setting?
The CAR-T cells currently approved are to be administered after lymphodepleting chemotherapy, generally consisting of cyclophosphamide and fludarabine, as this improves CAR-T cells expansion and persistence.
CAR-T cells are formulated as a dispersion/suspension for infusion. While, upon manufacture, these autologous products are promptly administered to the patient, they are cryopreserved. In the clinical practice, the CAR-T cells should be administered just after thawing.
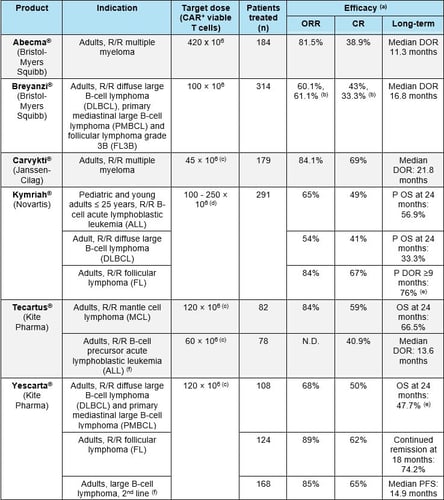
CAR-T cells are generally administered at a target dose in the approximate range of 50-400 x 106 CAR+ viable cells for an adult patient (Table 1). Dose adjustments are often recommended based on body weight.
Upon infusion, CAR-T cells expand rapidly, with median time of maximal expansion in peripheral blood at 7-15 days. CAR-T cells generally decrease to background levels by month 3, but in some cases CAR-T cells can be detected in peripheral blood for up to 2 years post-infusion. Importantly, transgene levels are positively associated with tumor response.
Unfortunately, serious adverse reactions are generally observed in approximately 50% of the patients. The most common serious adverse reactions include cytokine release syndrome (CRS), neutropenia, encephalopathy (immune effector cell-associated neurotoxicity syndrome, ICANS) or infection. Specific procedures are recommended for the management of these adverse reactions, including administration of tocilizumab and corticosteroids.
Which Indications Have Been Targeted?
CAR-T cell products are currently approved for the treatment of hematological malignancies, including different types of:
- leukemia
- lymphomas
- multiple myeloma (Table 1).
Treatment of solid tumors with CAR-T cells has been more challenging, mainly due to the histopathological characteristics of these tumors, the lack of tumor-specific antigens and the immunosuppressive tumor microenvironments (TME). Currently, no CAR-T cell products are approved for solid tumors, however multiple clinical studies targeting these conditions are ongoing, including glioblastoma, colorectal, pancreatic, gastrointestinal, thoracic or ovarian cancer 1.
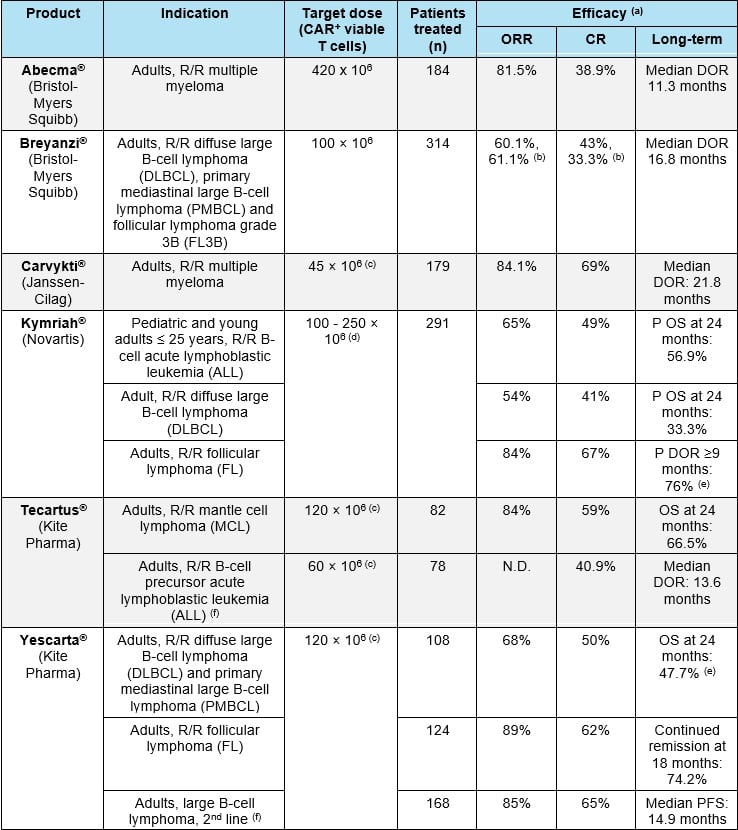
Abbreviations: CR: complete response; DOR: duration of response; ORR: overall response rate; P: probability; R/R: refractory/relapsed. (a) In all patients who underwent leukapheresis; (b) At the time of the most recent data cut-off (04 January 2021), with a median follow-up time of 11.6 months; (c) estimated for a 60kg person; (d) For patients 50 kg and below: 0.2 to 5 x 106 CAR+ viable T cells/kg body weight; for patients above 50 kg: 0.1 to 2.5 x 108 CAR+ viable T cells (non-weight based); (e) Median DOR: not reached. (f) Indication approved only in the US. References: European SmPCs and FDA labels for Abecma®, Breyanzi®, Carvykti®, Kymriah®, Tecartus® and Yescarta®.
Relapsed and Refractory Malignancies
CAR-T cells have initially been approved in relapse/refractory malignancies. However, it is expected that with the increasing data supporting the positive benefit-risk ratio of these therapies, upper lines of treatment will be pursued. Interestingly, the European draft Guideline on the clinical evaluation of anticancer medicinal products introduces the concept of "window of opportunity" for change, stating:
"Under certain well-defined conditions it is acceptable to conduct a clinical study with an experimental compound in settings (line of therapy, stage, etc.) where available data for this compound normally would be regarded as too limited (…)"
This "window of opportunity" may be applicable for Yescarta, which has recently been approved as a second line therapy for the treatment of patients with large B cell lymphoma (LBCL) by the FDA. Moreover, an ongoing Phase 2 study (NCT03761056, ZUMA-12) evaluates Yescarta as part of first-line treatment in 40 patients with high-risk LBCL 2. Therefore, it is likely that Yescarta may be indicated as a first-line therapy for the treatment of patients with LBCL in the near future. In such case, this would constitute a pivotal milestone in the shifting landscape of CAR-T cell therapies being developed as first-line therapies.
How Are Pivotal Studies Designed?
CAR-T cells have mostly been approved based on single arm studies, with efficacy discussed in the context of historical data with other therapies in the same patient populations. However, it is expected that as CAR-T cells progress to upper lines of treatment, randomized studies with active comparators will be needed. Indeed, the pivotal study for the indication of Yescarta as second line therapy for patients with LBCL (ZUMA-7) was a randomized study where patients were randomized to receive Yescarta or standard therapy.
In this line, it is worth noting that while the use of single arm studies for the treatment of relapse/refractory malignancies can be well justified, regulators strongly recommend randomized pivotal studies to demonstrate efficacy, including those studies using cell and gene therapies, as it is the case of CAR-T cells 3,4.
How Has Efficacy Been Measured in the Pivotal Trials?
The primary efficacy endpoint in pivotal studies for approved CAR-T cell products has been overall response rate (ORR). ORRs in the range of 50-80% were found in the different indications studied. These values are generally much higher than those obtained with standard treatments for the same indications (Table 1). Long-term efficacy has generally been measured as duration of response (DOR) or overall survival (OS, either real or modelled). Median DORs in the range of 10 - 22 months have been observed. Also, values of OS at 24 months are often determined.
While ORR has been considered a valid primary endpoint to demonstrate efficacy in the approved CAR-T cells, as they progress to upper lines of treatment and expand to more general indications, it is expected that more persuasive outcomes of efficacy, like OS or progression/disease free survival (PFS/DFS) 4 will be used as efficacy outcomes more frequently. When thinking about reimbursement and market access, it should be noted that assessments which correlate directly to clinical effectiveness are strongly preferred.
Which Are the Main Regulatory Guidance Documents?
The development of gene and cell therapy products (referred to as advanced therapy medicinal products, ATMPs, in Europe) has always been powered by intensive research in academia and hospitals. This has constituted a big challenge for regulatory agencies, as genetics and cell biology became a new paradigm to be understood and regulated.
However, in recent years, both the FDA and the EMA have released multiple guidance documents that constitute a sound basis for the development of gene and cell therapy products and to drive discussions with regulators throughout product development. (For a detailed list of these guidelines, please visit the corresponding pages in the FDA and the EMA websites).
Specific guidance for the development of CAR-T cells is provided in the Annex I of the 'Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells 3 and in the FDA draft Guidance for the Industry Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products 5. Additional relevant recommendations are outlined in the guidelines for the clinical development of oncological products, as published by the FDA and the Evaluation of anticancer medicinal products in man - Scientific guideline.
To Conclude
Although the clinical development of CAR-T cells is very challenging, a great deal of experience has been accumulated during the development of the currently approved CAR-T cell products and their subsequent use in the clinical setting. Reviewing the successful development of the approved products can be very useful for the future development of new CAR-T cells or new indications, as well as for similar products in the oncology field, including CAR-NK, engineered TCR cells or TILs. However, development plans should take into account that higher expectations for upper treatment lines may need to be addressed. In addition, multiple guidance documents published by the EMA and the FDA provide fundamental guidance for the development of gene and cell therapy products, and outline what the current expectations of the regulatory agencies are. These guidance documents should be taken into account in discussions with the regulatory authorities to expedite the approval of such products.
ProPharma holds significant expertise within development of cell and gene therapies and is uniquely positioned to help you navigate this field, including CAR-T cells and many other related innovative products (allogenic CAR-T cells, CAR-NK cells, TILs, etc.). At ProPharma, we are continually reviewing the landscape to maintain current knowledge of the latest developments, technologies, innovations and regulations within the cell and gene therapy realm to best support our Clients in the scientific discussions with the regulatory authorities and streamline the development and approval of the most innovative products in the cell and gene therapy horizon.
Interested in learning more? Contact us today to find out how we can help with your global regulatory needs.
References
- Patel, U. et al. CAR T cell therapy in solid tumors: A review of current clinical trials. eJHaem 3, 24-31, doi:https://doi.org/10.1002/jha2.356 (2022) ↩ Back to reference
- Neelapu, S. S. et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: the phase 2 ZUMA-12 trial. Nature Medicine 28, 735-742, doi:https://doi.org/10.1038/s41591-022-01731-4 (2022). ↩ Back to reference
- EMA/CAT/GTWP/671639/2008 Rev. 1 - corr. Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells ↩ Back to reference
- EMA/CHMP/205/95 Rev.6. Guideline on the clinical evaluation of anticancer medicinal products - draft, (2019). ↩ Back to reference Back to reference
- Center for Biologics Evaluation and Research (CBER). Chimeric Antigen Receptor (CAR) T Cell Products - Draft Guidance for Industry (2022).↩ Back to reference

