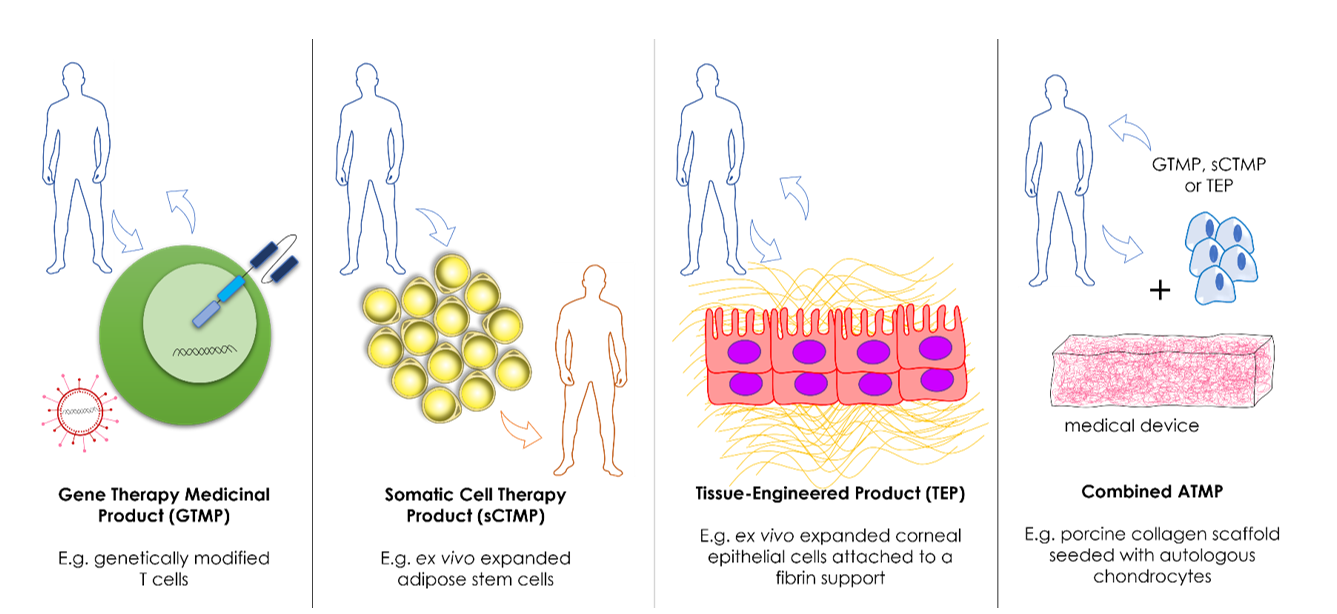
We are living through a medical revolution. Advances in gene therapy, cell‑based therapies and tissue engineering offer real hope for patients with a range of debilitating diseases.
The FDA, EMA and other global regulatory agencies are still learning how to evaluate the benefits and risks of these innovative products. Each case is unique, and often many uncertainties remain at the time of licensing.
How should sponsors prepare their regulatory strategy? Using our years of experience, and understanding the inherent regulatory uncertainties, we suggest five keys to success.
Key 1: Create a truly integrated team
For advanced therapies, the interplay between manufacturing, quality control, non-clinical and clinical development is far more extensive than for conventional drugs or biologics. Development teams that communicate effectively with each other will better understand the inter-dependence of each discipline. They will plan the development program more accurately, and in doing so, build trust with the regulators.
For example, an apparently subtle change to cell culture media or introduction of automation during scale-up could dramatically alter the performance of a cell‑based product in animal models and in the clinic. This might occur even if analytical comparability has been demonstrated using in vitro assays. Is the clinical team aware of the timelines and potential impact of every manufacturing change being planned during the trial program? A highly functioning, integrated team will be able to develop well justified, risk-based bridging strategies, and present timelines relative to the BLA or MAA submission date. This will give regulators maximum visibility of the path ahead and help to avoid surprises late in development.
Key 2: Engage the regulators as partners
The most successful sponsors approach the regulators as partners to be engaged with, not adversaries to be overcome. Early and transparent dialogue with the agencies will foster their support and may uncover alternative paths to generate the evidence needed for product quality and benefit-risk evaluation. Sponsors should disclose critical gaps before the pre-BLA / pre-MAA stage, so there is still time to implement solutions that best reassure the regulators.
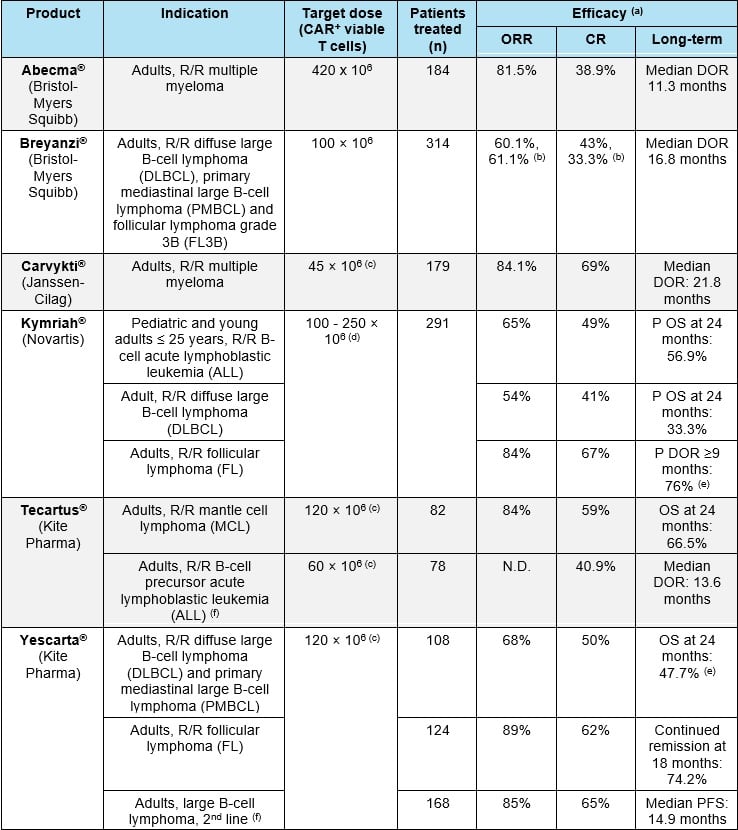
Every R&D program has its gaps, and this is particularly true for gene and cellular therapies. Relevant animal models for pharmacology and toxicology are often unavailable due to species differences, and the product’s mechanism of action and off-target effects must be explored in vitro and in patients. It may be necessary to develop novel potency tests, rapid release tests, biomarkers or clinical endpoints that have no regulatory precedent. And for rare diseases, comparison with historic or real-world data may be needed in place of large randomized trials, as exemplified recently by the SCHOLAR-1 dataset that supported the approval of Yescarta.
The regulators can sometimes bring a fresh perspective to these issues from their experience with other sponsors and share their latest thinking. The FDA’s INTERACT program and the EMA’s Innovation Task Force offer opportunities to receive guidance early in development programs, mitigating unnecessary expenditure. The time and effort invested in preparing for agency meetings is valuable in itself, allowing a busy development team to evaluate the path ahead and identify rate limiting activities.
Key 3: Invest in regulatory intelligence
Some regulatory requirements may appear conservative, because the regulators are accountable for patient safety. However, a good understanding of the agencies’ default position and their previous judgments will allow sponsors to justify the exceptions that may be needed for their product.
As their experience increases, FDA and EMA have released a series of guidelines to address specific topics in the development of gene and cellular therapies (also known as ‘advanced therapies’ in Europe). Insight into the agencies’ thinking can also be gained from speakers at conferences and multi-stakeholder forums. Important learnings can also be gained from careful examination of precedents. As illustrated in Table 1, a number of advanced therapies have gained regulatory approval in the US, Europe or Japan in recent years, and assessment reports are publicly available.
Table 1: Recent BLA / MAA outcomes for gene, cell and tissue therapies, 2016-2019
| Product |
Description |
Regulatory outcome |
| Zynteglo |
Cell based GT for beta-thalassemia |
Approved in EU 2019 |
| Raligeze |
GT for cervical cancer |
Application withdrawn in EU 2018 |
| Alofisel |
Stem cell therapy for rectal fistula |
Approved in EU 2018 |
| Kymriah |
CAR T cell therapy for DLBCL, ALL |
Approved in US 2017, EU 2018 |
| Yescarta |
CAR T cell therapy for DLBCL, PMBCL |
Approved in US 2017, EU 2018 |
| Luxturna |
GT for retinal disease |
Approved in US 2017, EU 2018 |
| Spherox |
TEP for cartilage diseases |
Approved in EU 2017 |
| Zalmoxis |
Stem cell therapy as HSCT adjunct |
Approved in EU 2016 |
| Strimvelis |
Cell-based GT for SCID |
Approved in EU 2016 |
| Jace |
TEP for severe burns |
Approved in Japan 2016 |
ALL = acute lymphoblastic leukemia; CAR = chimeric antigen receptor; DLBCL = diffuse large B cell lymphoma; GT = gene therapy; HSCT = hematopoietic stem cell transplant; PMBCL = primary mediastinal B cell lymphoma; SCID = severe combined immunodeficiency disease; TEP = tissue engineered product
Key 4: Remember that products and portfolios evolve
Given the rapid progress of technology, the post-approval lifecycle management of an advanced therapy will be more complex than for a conventional drug or biologic. Sponsors should bear in mind the future evolution of their product when planning the initial BLA or MAA.
For instance, a chimeric antigen receptor (CAR) T cell therapy might be modified with a new culture medium, the addition of a genetic suicide switch to inactivate the product in case of life-threatening side effects, or switching to a decentralized manufacturing model where the product is produced in the local hospital. Depending on the nature of each proposed change, the regulators may consider them to constitute a new product, with all the evidence generation that this implies. Transparent discussion with regulators, and appropriate implementation of their recommendations, can effectively anticipate such complications in the lifecycle management of the product.
Key 5: Think globally
For good economic and regulatory reasons, sponsors generally focus on the United States as the site of product manufacture, and for the initial product launch. A global regulatory strategy typically emerges only during late phase development. However, the complexity of advanced therapy regulations in each region demands a more proactive approach. In particular, local regulations will significantly impact plans for regional manufacturing, transport and storage of human materials, data privacy, and the evidence needed to support reimbursement.
If the product is manufactured in a different geography to the patient, a host of rules on import, export and data privacy need to be considered, not to mention the validation of handling, storage and shipment. Navigating these requirements takes a great deal of planning and is especially critical for supply chains that rely on rapid transport of chilled tissue samples, or for autologous products for which robust traceability is essential for patient safety. Advice meetings with the regulators are essential to understand their expectations beyond the published guidelines.
Opening a clinical trial in Europe or Japan may be complicated for products containing genetically manipulated cells, which fall under the scope of Genetically Modified Organism (GMO) laws that were originally designed for agriculture. In Europe, GMOs are regulated at a country level rather than by EMA, and in some countries the process can delay clinical trial startup by many months. The Cartagena Act in Japan can have a similar impact.
In Europe, the so-called ‘hospital exemption’ provides the possibility for hospitals to manufacture their own therapies for individual patients without needing a marketing authorization, although GMP rules still apply. Understanding the potential competitive impact of this exemption in each market will be important for sponsors wishing to introduce a conventionally licensed product. Not surprisingly, this is currently a hot topic in EU regulatory policy.
For rare diseases, Europe sets a high hurdle for maintenance of orphan designation at the time of marketing authorization. To avoid losing orphan status, additional data generation and quantitative analysis is needed to demonstrate ‘significant benefit’ versus all other available treatment methods. Early discussion with EMA may improve the chances of maintaining the orphan designation, which in turn impacts pricing and market access.
Finally, advanced therapies are also often eligible for accelerated / conditional regulatory pathways, which are best discussed before initiating the pivotal trial(s). The authorities in Europe, Japan and elsewhere may not accept the same post-marketing studies and long-term follow-up that was agreed with the FDA, and they may require substantially different risk management plans. It becomes far more challenging to negotiate and/or prepare for these issues as the MAA approaches. For this reason, a pro-active global regulatory strategy is essential.
Developing a gene, cellular or tissue engineered therapy is a complex business, with many uncertainties along the way. But sponsors who follow these five keys to success may enjoy a smoother path to regulatory approval.
ProPharma Group, incorporating ProPharma Group and Southwood Research, has years of experience engaging with the FDA and EMA. We are well placed to advise sponsors on development and preparation of the global regulatory strategy, successful meetings with the regulators, as well as assisting with CMC, non‑clinical, clinical and quality compliance throughout the development program. If we could assist you, please contact ProPharma Group, a ProPharma Group Company.