August 26, 2015

August 26, 2015

In the previous installment of this blog, we discussed the importance of establishing a Master Validation Plan, and how to plan and assess your validation approach. Today’s segment will complete the discussion, focusing on calculating the degree of risk for each instrument, assessing documentation, selecting team members, and developing a schedule of deliverables.
+Risk
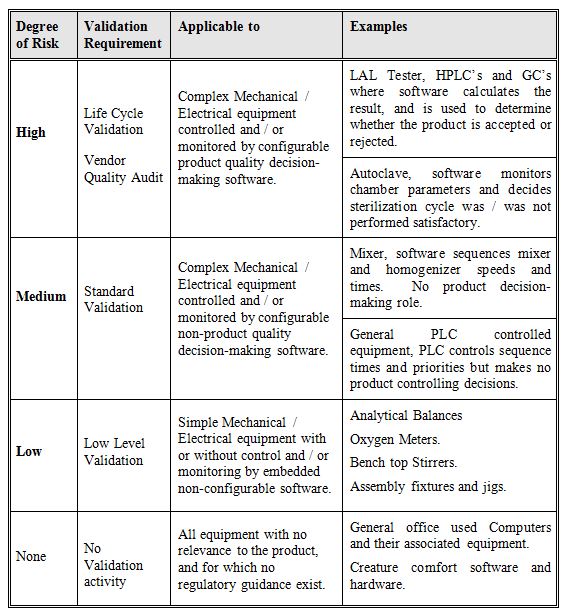
Calculate the degree of risk for each instrument, four possible categorizes are given in table 1 below, one being, ‘No Validation’ the other three levels of validation have been chosen as being sufficient to ensure that the equipment under qualification will receive the appropriate level of validation.
Table 1: Degree of Risk
+ Documentation
It is also important to assess the documentation system associated with your qualification project. In general, all governing SOPs should be assessed to ensure the project will proceed in an efficient and compliant manner. SOPs associated with the equipment must also be updated and a clear plan for defined roles and responsibilities should be set. For example, the defined role and responsibilities for the development, review, and approval of the SOPs should include the roles (as applicable) listed below – with a brief description for each and the responsibility (in a general way) as it applies.
Roles and Responsibilities
1. Management level – Responsible for reviewing and approving SOPs
2. Quality Assurance – Responsible for reviewing and approving SOPs
3. System Owner – Responsible for reviewing and approving SOPs
4. Operations – Responsible for determining required SOPs, providing developmental support for SOPs, reviewing and approving SOPs
5. Technical and Engineering support – Responsible for determining required SOPs, providing developmental support for SOPs, reviewing and approving SOPs
6. Validation Specialist – Responsible for determining required SOPs, providing developmental support for SOPs, reviewing and approving SOPs
7. Technical Writers - Responsible for developing SOPs
+ The Team
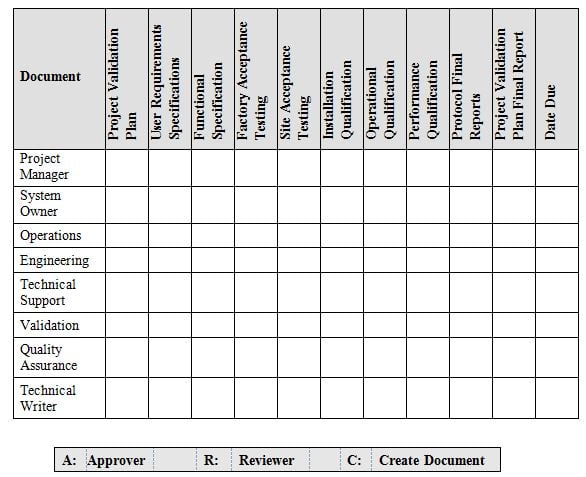
It is now evident that the qualification program cannot be handled by any one individual. Its going to require a Team effort to assure compliant execution of your qualification program. The team members should be selected from various departments indicated in table 2 below. The list of Team members is not all inclusive, but is an example of various departments at a minimum that should be included.
The Team will be responsible for timely review and approval of protocols for their respective area of expertise. Additionally they will address any deviations, corrective and preventative actions, procedure development, instrument scope and changes, resource requirements and any other qualification topics that may arise and affect the deliverable timeline.
+ Deliverables
A description of the overall schedule for validation of the instrumentation should be supplied, with indication of the planned end dates for the validation milestones in the project (see table 2 below). This timeline is intended as a preliminary baseline for the project. A detailed analysis of resource allocation, activities and timing would usually be given in the plan.
Table 2
In conclusion, there is much to consider about your Analytical Instrument Qualification program prior to embarking on implementation. Remember, prepare a validation master plan and/or equipment qualification plan, implement an assessment approach that encompasses risk priority for each analytical instrument, a comprehensive historical list of all documentation associated with the instrument, a time line for deliverables, and finally assemble a qualified team to execute the qualification protocols. When all these elements are addressed, you can safely answer, yes, I am ready for AIQ!
TAGS: Analytical Instrument Qualification Life Science Consulting